Molecular Genetics
Jonathan Flint
Introduction
The transformation of the LOD score (an acronym for log of the odds ratio), from obscurity as a footnote in medical genetics, to celebrity as multiple choice test item in professional examinations in psychiatry, epitomizes the invasion of genetics, and particularly molecular genetics into psychiatric research. Moreover, like other celebrities caught up in fast moving fields, LOD scores are likely to return to their humble origins within a few years. As molecular genetic approaches to mental health move away from simply identifying genes and DNA sequence variants towards functional studies of increasing complexity, newcomers to the field have to master an expanding literature that covers diverse fields: from quantitative genetics to cell biology, from LOD scores to epigenetics. This chapter takes on the task of making the reader sufficiently familiar with the broad range of subjects now required to follow the progress of psychiatric genetics in the primary literature.
A number of achievements have to be highlighted. Foremost among these is the completion of the human genome project. Announced annually from 2001(1, 2 and 3) and thereby begging the question as to what constitutes completion, the human genome project is now an essential biological resource. As expected, the ability to sequence whole genomes has transformed the way genetics is carried out, perhaps most egregiously with the rise of bioinformatics as a core discipline: discovery now takes place using the internet rather than the laboratory. Anyone with an interest in human biology should look at the frequently updated information at http://www.ensembl.org or http://genome.ucsc.edu.
Without the human genome two other critical developments would have been impossible: the ability to analyse the expression of every gene in the genome and the ability to analyse (theoretically at least) every sequence variant. Both developments also depend on miniaturization technologies that enable the manufacture and interrogation of initially thousands and then millions of segments of DNA. In addition, results from the International Haplotype Map (HapMap) project,(4) which catalogues common variation in the human genome have been crucial in making it possible to take apart the genetic basis of common, complex disorders such as depression, schizophrenia, and anxiety.
Few disciplines are more burdened with jargon than molecular genetics. This is partly due to the proliferation of molecular techniques, but it is also partly intrinsic to the subject; the only unifying principle is evolution, which often operates in a very ad hoc fashion. Biological solutions to the problems posed by selection result in the adaptation of existing structures to new uses, rather than to the invention of purpose-built systems. Consequently there are few general lessons to be learnt and the novice simply has to become adept at recognizing the acronyms and neologisms that decorate the literature. The material in this chapter aims to equip the reader with the necessary terminology. It begins with the structure and function of DNA, an essential starting place for a number of reasons.
Nucleic acid structure and function
The chemical constituents of genetic information are deoxyribonucleic acid (DNA) and ribonucleic acid (RNA). Both molecules consist of linear chains of nitrogenous bases bound to a sugar (ribose) and a phosphate backbone. Because of the way the sugars are joined together, one end of each nucleic acid strand will have a terminal sugar residue in which the carbon atom at position number 5 of the ribose molecule is not linked; the other end has a free carbon atom at position 3. These two ends are termed the 5′ (5 prime) and 3′ (3 prime) ends respectively.
It is usual to describe a DNA or RNA sequence by writing the order of bases in a single strand, in the 5′ to 3′ direction. DNA contains four nitrogenous bases: adenine (A), guanine (G), cytosine (C), and thymine (T). RNA differs in that it contains uracil instead of thymine.
Two structural differences between DNA and RNA are important for understanding nucleic acid function. First, DNA has a hydroxyl group on part of its sugar constituent whereas RNA has a hydrogen atom. The result is that, in most biological environments, RNA is much more unstable than DNA. Second, RNA normally exists as a single molecule, whereas DNA is a double helix in which two strands are held together by weak hydrogen bonds between opposed base pairs (bp), C joined to G and A to T. The sequence of one strand can therefore be inferred from the other. The two strands are said to be complementary to each other, and this property is exploited whenever DNA is copied (during meiosis, mitosis, or in vitro processes such as amplification of DNA using a polymerase chain reaction).
As befits an unstable molecule, RNA mediates the expression of genetic information; its production and degradation are tightly controlled. RNA is translated into a linear order of amino acids in proteins according to a three-letter code (e.g. GAA encodes the amino acid glutamine). DNA acts as a template for the production of RNA in a process termed as transcription. But DNA is more than a stable repository of encoded protein sequence information; it also contains information that controls the transcription of RNA.
Disorders of the template function of DNA are the molecular basis of inherited dispositions and illness, and are the subject matter of genetics. By contrast, gene expression (the transcription and translation of RNA) is not entirely genetically predetermined. It is highly regulated, but in response to changes in the cellular environment which in turn reflect changes in the state of the organism. Disorders of gene regulation are now emerging as important causes of disease.
Genome organization
DNA within cells is packaged into chromosomes in the cell nucleus, with a tiny amount (16 569 bases containing 37 genes) in the mitochondria. Since mitochondria in the fertilized egg are maternally derived, mitochondrial inheritance is through the female lineage. Although small, mitochondrial disorders contribute substantially to degenerative disorders including ageing. More important is nuclear DNA.
As of 2007, the size of the nuclear genome is 3 253 037 807 bp (3.3 gigabases) containing 21 662 known and 1064 novel genes (http://www.ensembl.org). Packing such a large molecule into a cell is done at a number of levels, with profound consequences for gene function. At the first level, 147 bp lengths of DNA are wrapped around octamers of proteins known as histones. These nucleosomes are the fundamental units of the state of packaged DNA known as chromatin.
Packaged DNA is itself organized into 22 autosomal chromosomes, one inherited from the mother and one from the father, and two sex chromosomes, X and Y. Each chromosome pair exchanges stretches of DNA during sexual division (meiosis) in a process called recombination (without which genetic mapping, the basic method of finding disease genes, would be impossible). Chromosomes have three functional elements: origins of replication, centromeres, and telomeres. Replication origins are required to initiate DNA replication and maintain chromosome copy number. Their molecular structure is unknown. Centromeres are responsible for the segregation of chromosomes during cell division. Their molecular nature is also not understood, but they are visible in light microscopy as a constriction where the duplicated chromosomes (called chromatids) are held together. Chromosomes are said to have two arms, separated by the centromere which, despite its name, is not always at the centre. Short arms are termed p (petit) and long arms q (queue). Telomeres are the ends of chromosomes and their molecular nature is well understood. They consist of long stretches of the sequence TTAGGG without which the chromosome is unstable, tending to break apart and fuse with itself or to other chromosomes.
No one has found any general principles that organize genetic material within chromosomes. Rather than being an efficiently organized plan of the organism consisting of precise drawings, the genome resembles a working copy written over countless rough drafts and discarded versions, among which there are literally thousands of jottings and scribbles, most irrelevant to the final structure. While there are examples of gene families clustered in the same chromosomal location (for instance genes involved in immune regulation are clustered on chromosome 6p), more commonly the position of genes on chromosomes does not reflect functional similarity. For example, genes expressed only in one tissue, or at one stage of development, are often immediately adjacent to widely expressed housekeeping genes.
Genes and the regulation of gene expression
DNA is transcribed into RNA, which in turn is translated into protein. Transcription involves the excision of large portions of transcribed RNA (by RNA splicing enzymes) and modifications to the ends of the RNA molecule (capping of one end and polyadenylation of the other). The final product, messenger RNA (mRNA),
contains a central section, translated into protein, and flanking non-coding regions. The consequence of these manipulations is that DNA and mRNA are not coterminous; sections of DNA that encode mRNA (termed exons) are interrupted by often very large stretches of DNA that are not translated (termed introns) (Fig. 2.4.2.1).
contains a central section, translated into protein, and flanking non-coding regions. The consequence of these manipulations is that DNA and mRNA are not coterminous; sections of DNA that encode mRNA (termed exons) are interrupted by often very large stretches of DNA that are not translated (termed introns) (Fig. 2.4.2.1).
Gene expression is controlled in a number of ways, predominantly at the level of transcription (but posttranscriptional processing and translational control are important for some genes). While knowledge in this area is still rudimentary, but advancing fast, it has already led to new insights in disease aetiology: understanding theories of the neurobiology of depression now requires familiarity with chromatin remodelling; epigenetic effects are often invoked in molecular biology of intellectual disability. Below I summarize information about the relevant molecular processes.
1. Transcription factors: transcription factors exercise control over gene expression. Transcription occurs when RNA polymerases manufacture RNA from the template DNA, a process that requires the help of transcription factors, proteins that recognize and bind to specific DNA sequences (note that transcription factors can also repress transcription). Although transcription factor-binding sites are found close to a gene, at a 5′ region known as the promoter (see Fig. 2.4.2.1), they may also be situated far away, sometimes within other genes. The characterization of these control elements remains a major challenge for genome research and is currently a focus of the ENCODE project, a continuation of the human genome project whose aim is the comprehensive identification of all the functional elements in our genome (http://genome.ucsc.edu/ENCODE/).
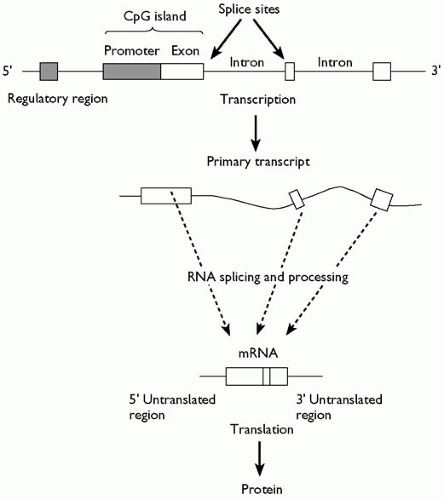 Fig. 2.4.2.1 At the top of the figure the organization of a gene in genomic DNA is shown. Unshaded boxes correspond to coding regions (exons) and the two shaded boxes correspond to control regions. The control region immediately 5′; of the first exon, where transcription is initiated, is known as the promoter and often has a characteristic sequence composition. In almost all ubiquitously expressed genes (and in many tissue-specific genes) it is unmethylated, GC rich, and has a relative excess of the dinucleotide CpG. The region, which typically contains the first exon as well as the promoter, is called a CpG island. The boundaries between exons and introns are called splice sites and are conserved; introns virtually always start with the sequence GT and end with the sequence AG. The entire genomic region is transcribed into a primary transcript (bold arrow) which is then processed to excise the introns. Many human genes undergo alternative splicing to yield a number of different mRNA products. Mature mRNA is then translated into a protein product. |
Transcription factors typically control the expression of a number of genes, reflecting the presence of a hierarchical structure of coordinated gene expression. Consequently mutations in transcription factors have effects on different, seemingly unrelated phenotypes, a phenomenon called pleiotropy. The constellation of phenotypic abnormalities seen in intellectual disability syndromes can be explained in this way. For example mutations in the X-linked ATRX gene result in an anaemia (α-thalassaemia), a characteristic facial appearance, profound developmental delay, neonatal hypotonia, and genital abnormalities.(5) The gene contains sequence motifs indicating that it belongs to a group of proteins that bind to chromatin and is involved in chromatin remodelling, discussed below.(6)
2. DNA methylation: gene inactivation is associated with DNA methylation, predominantly the addition of methyl groups to cytosine bases. DNA methylation occurs almost always at CpG dinucleotides, and most CpGs in the genome are methylated.(7) DNA methylation represses gene expression in two ways: first, modification of cytosine inhibits the association of some DNAbinding factors with their DNA recognition sequences, and second proteins that recognize methyl-CpG can repress transcription from the methylated DNA. Methylation of DNA does not change the DNA sequence itself, as it is reversible, but the methylation status is maintained when cells divide. Consequently the change is referred to as epigenetic modification.
DNA methylation is critical for imprinting, a form of gene regulation in which transcripts are expressed from only one of the two parental chromosomes. Although there are relatively few imprinted genes (about 60 are currently documented, although sequence features in the region of known imprinted promoters implicate 600) imprinting is an important phenomenon in neurobiology for three reasons(8): (i) There is evidence from studies of embryos that maternally expressed genes enhance and paternally expressed genes reduce brain size, indicating that at least some imprinted genes are likely to be involved in neurodevelopment.(9) (ii) Disorders of imprinting are important in intellectual disability syndromes: Rett syndrome, Prader-Willi syndrome, Turner syndrome, and Angelman syndrome.(10) (iii) Imprinting is involved in X chromosome inactivation, the mechanism by which cells compensate for males having just one copy of the X chromosome while females have two.(11) The X chromosome contains a disproportionately high density of loci affecting cognition and since males always inherit their X chromosome from their mother, the presence of X-linked imprinted genes is believed to contribute to sexually dimorphic effects.
The biology of imprinting is complex and not fully understood. In germ cells and in pre-implantation embryos, all methylation
patterns are removed, and then re-established.(12) About half of identified imprinted genes are clustered within imprinting centres (IC) which carry the allele-specific methylation marks established at this developmental stage. Most maternally silenced imprinted genes are repressed by promoter methylation. No protein-coding imprinted gene has been found that is repressed by paternal methylation derived from the sperm (presumably because of active demethylation of the paternal genome). Importantly, paternal repression is achieved, at least in some cases, by using a transcript on the opposite strand (an antisense transcript). Promoter methylation of the antisense transcript (usually resulting from oocyte-derived methylation) represses its transcription and thus activates the protein-coding gene epigenetically.
patterns are removed, and then re-established.(12) About half of identified imprinted genes are clustered within imprinting centres (IC) which carry the allele-specific methylation marks established at this developmental stage. Most maternally silenced imprinted genes are repressed by promoter methylation. No protein-coding imprinted gene has been found that is repressed by paternal methylation derived from the sperm (presumably because of active demethylation of the paternal genome). Importantly, paternal repression is achieved, at least in some cases, by using a transcript on the opposite strand (an antisense transcript). Promoter methylation of the antisense transcript (usually resulting from oocyte-derived methylation) represses its transcription and thus activates the protein-coding gene epigenetically.
Some evidence, obtained by examining females with a single X chromosome (Turner syndrome females), indicates that imprinted genes on the sex chromosomes influence brain structure and function.(13) Since the single X chromosome is inherited from either the mother or father, it is possible to compare the effects attributable to a maternally or paternally imprinted chromosome. Maternally expressed X-linked genes have been reported to influence hippocampal development, while paternally expressed genes influence the normal development of the caudate nucleus and thalamus in females. Using Turner syndrome patients, Skuse and colleagues have suggested that a paternally expressed allele is associated with enhanced social-cognitive abilities.(14) A similar observation has been found in a mouse model of Turner syndrome, lending weight to the view that imprinted genes are involved in cognitive processes.(15)
3. Chromatin remodelling: the nucleosome forms a barrier to transcription, primarily because DNA has to be free of nucleosomes for it to be accessible to transcription factors and the large complex of proteins that constitutes RNA polymerase. To some extent the organizational information of where nucleosomes are positioned is embedded within DNA sequence, in a nucleosome code; but the nucleosome is not a static unit. It too has dynamic properties and exerts an effect on transcription. Furthermore, like methylation, the effects are heritable, providing a second form of epigenetic modification to DNA (X inactivation also involves this form of epigenetic modification).
Histones (the proteins that constitute nucleosomes) are subject to a large number of modifications (acetylation, ubiquitination, methylation, ADP-ribosylation, and sumolation of lysine residues; and phosphorylation of serines and threonines) of which two, lysine acetylation and methylation, have been most heavily studied. Histone modifications can influence each other and may also interact with DNA methylation, in part through the activities of protein complexes that bind modified histones or methylated cytosines. These, and other proteins that remodel chromatin, control genes involved in the development and activity of the central nervous system.
Chromatin remodelling has attracted attention as a possible mechanism for bringing about persistent change subsequent to an environmental stressor.(16) Two examples are relevant. Meaney and colleagues have reported that heritable differences in stress reactivity in rats depend on variation in parenting, not variation in DNA.(17) Adult offspring of mothers that show higher levels of licking, grooming, and arched-back nursing (high-LG-ABN) are less fearful and show more modest hypothalamic-pituitary axis responses to stress than offspring of ‘low-LG-ABN’ mothers.(18) How are these maternal effects, or other forms of environmental programming, sustained over the lifespan of the animal?
Variations in maternal care were found to alter the methylation status of a promoter of the glucocorticoid receptor gene. Central infusion of a histone de-acetylase inhibitor enhanced histone acetylation of the glucocorticoid receptor promoter in the offspring of the low-LG-ABN mothers. Analysis of the promoter showed that CpG dinucleotides were hypomethylated. In consequence, the maternal effect on hippocampal glucocorticoid receptor expression and the hypothalamic-pituitary axis response to stress were both eliminated.(19) This finding suggests that there is a causal relation between epigenetic modifications, glucocorticoid receptor expression, and the maternal effect on stress responses.(20)
Nestler’s group invoked chromatin remodelling as an explanation for the long-lasting behavioural change induced by antidepressants.(21) Chronic defeat stress in rodents is reversed by chronic (but not acute) antidepressant treatment, a model for the action of antidepressant action in our own species. Chronic defeat stress and chronic antidepressant treatment are associated with reciprocal, long-lasting changes in expression levels of brain derived neurotrophic factor (BDNF). This is in turn associated with lasting changes in chromatin architecture at the corresponding BDNF gene promoter. Furthermore, down-regulation of a histone de-acetylase (Hdac5) by chronic antidepressant treatment was necessary for the therapeutic efficacy of the antidepressant.
4. Small RNAs: small RNAs include micro RNAs (miRNAs) and small interfering RNAs (siRNAs) directly or indirectly alter gene transcription.(22, 23) siRNAs, derived from double-stranded RNAs (dsRNAs), control cleavage of other transcripts and can themselves direct the production of dsRNA by RNA-dependent RNA polymerase; they are also implicated in recruiting heterochromatic modification that leads to transcriptional silencing.
The extent of siRNA involvement in eukaryotic gene regulation is still unclear. Micro RNAs specify posttranscriptional gene repression by base pairing to the messages of protein-coding genes. They represent nearly 1-5 per cent of all genes in higher eukaryotes and have been implicated in developmental timing and neuronal patterning. They are believed to facilitate the transition between developmental stages and therefore are likely to have an effect on the expression and evolution of most mammalian mRNAs.
Only 3 per cent of the genome codes for protein.(24) However, that is not to say that the remaining 97 per cent is inactive; in fact the number of transcripts is at least 10 times as great as the number of genes.(25,26) What this means in terms of function is not clear, but along with the discovery of small RNAs it has forced a re-evaluation of what is meant by a gene. The idea that a gene is a section of DNA transcribed into RNA, which in turn encodes a protein, fails to capture the gamut of RNA species, some of known function (such miRNAs), some with only suspected function. The emerging complexity of gene function and the multiple species that need to be included in any definition of a
gene has dramatically increased our understanding of molecular pathogenesis.
gene has dramatically increased our understanding of molecular pathogenesis.
Genetics and genotyping
Chromosomes are not stable structures. They rearrange during meiosis, recombining material between the paternal and maternal chromosomes. The mechanisms of recombination are not relevant for understanding neurobiology, but without recombination we would not be able to track mutations. This section describes the basic methodologies currently used. To follow it, and the many reports in the literature, it is essential to be familiar with genetic terminology. A brief reminder is provided next.
A position on a chromosome is called a locus, a general term which can refer to a gene or a segment of DNA with no known function. DNA sequences that differ at the same locus are called allelic variants. Since we have two copies of each chromosome, by definition we have two alleles at each locus. If these alleles are identical the individual is said to be a homozygote; if they are different, the individual is a heterozygote. It follows that for a locus with two alleles (that is one which is di-allelic, as are the single nucleotide polymorphisms that form the basis of almost all genetic mapping experiments), then there are three possible genotypes. For example, if the alleles are either C or T, the possible genotypes are CC, CT, and TT, whose frequencies in a population, in the absence of migration, mutation, natural selection, and assortative mating, are a simple function of allele frequencies (this phenomenon is termed Hardy-Weinberg equilibrium).
The relationship between alleles at different loci is important for genetic mapping. Assume there are two loci on the same chromosome, separated by approximately 1 megabase (Mb). The loci are again di-allelic, the first with alleles C and T, the second with alleles A and G, so that there are nine possible two-locus genotypes: ATCC, TTCG, AAGG etc. Consequently, the chromosomes of an individual with the two-locus genotype ATCG could be any of the following four: A-C, A-G, T-C, or T-G. This combination of alleles along a chromosome is known as a haplotype. If recombination occurs between the two loci it will break-up the haplotype, so that the offspring of someone with the haplotype A-C on one chromosome and T-G on the other may inherit the novel haplotype A-G. The probability at which this occurs depends on the genetic distance between the two loci. For 1 Mb in the human genome this probability is approximately 1 per cent per meiosis, or 1 cM.
Molecular mapping depends on the availability of genetic markers across the genome. The genome is replete with DNA sequence polymorphisms whose only known use is to enable geneticists to map disease genes. On average, every 1000 bp will contain 1.4 bases that differ between two randomly chosen individuals, almost all of which have no phenotypic consequence. In addition, there are small runs of repeated sequence (most commonly CA) which differ in length between individuals. At least one of these short tandem repeats (STRs), or microsatellites, is found every 50 kilobases (kb) and they also have no known phenotypic consequences. There are other more complex sequence polymorphisms, but single nucleotide polymorphisms (SNPs) and microsatellites are the most useful for identifying disease genes.
Genotyping of genetic markers almost always starts by amplifying DNA using the polymerase chain reaction (PCR). PCR requires the following reagents: a DNA polymerase; a pair of oligonucleotides (also referred to as primers), which are synthetic single-stranded DNA, usually between 15 and 25 bases long, complementary to two sequences on opposite strands of the target DNA; the target DNA itself; all four nucleotides, usually present in excess; appropriate buffer and cofactors for the reaction. The reaction proceeds in a cycle of three steps: (1) the mixture is heated to over 90°C for 1 min to separate the complementary strands of target DNA, (2) the mixture is cooled, to about 50°C, so that the oligonucleotides anneal to their complementary sequence in the target DNA and allow the DNA polymerase to bind (oligonucleotides are required to prime the polymerase), and (3) the temperature is adjusted to allow the polymerase to function in the extension component of the reaction. Typically the polymerase is from a thermophilic bacterium with a permissive temperature of 72°C. Products from the first cycle serve as targets for a second round of amplification and so on, for up to about 40 cycles.
The method used to genotyping the PCR product depends on the nature of the sequence variant. Microsatellite genotyping involves discriminating the length of the PCR products, usually accomplished by separating the fragments by electrophoresis, in which an electrical current causes smaller fragments to migrate more quickly than larger fragments through a matrix. Typically a fluorescent label is incorporated into the amplified DNA allowing the PCR products to be detected by a laser, a method that allows automated analysis.
SNP genotypes can also be worked out from differences in molecular weight. For example, the nucleotide added to a primer complementary to sequence immediately preceding a SNP in the PCR fragment will depend on the genotype of the individual. The small difference between a primer with a C nucleotide added to its end (reflecting the presence of a G nucleotide in the PCR fragment) compared to the same primer with the addition of a T nucleotide, can be determined using a mass spectrometer. However, most high-throughput genotyping methodologies (required for whole genome association studies) exploit the specificity of DNA hybridization: while two complementary single strands of DNA stick (or hybridize) together (ATTGAC will anneal to TAACTG) a single base mismatch will prevent hybridization (ATTGAC will not anneal to ATAGAC). Consequently, a SNP can be detected by determining whether primers hybridize to the PCR fragment. By labelling the primers, for example, with a fluorescent dye, the hybridization can be visualized, and, using the same technology that builds computer semiconductors, millions of primers can be manufactured on a small piece of glass, allowing the simultaneous detection of millions of SNPs.
Genetic mapping: linkage and association
For genetic disorders that arise from a mutation in a single gene, behaving in a Mendelian fashion (dominant, recessive, or sex-linked inheritance), disease gene identification is conceptually straightforward. Marker alleles follow Mendelian laws of segregation so that it is possible to determine if a marker is co-segregating with a disease in a family and to test the result statistically using methods described in Chapter 2.4.1. The expected result is an estimate of the probability that an allele and a disease locus will recombine; the lower the probability, the closer together the two loci are on the chromosome. The statistical test gives the likelihood that the estimate of recombination distance between a marker and a mutation
is correct. If the likelihood is acceptably high, then the next task is to reduce the genetic interval as much as possible and to identify a causative mutation. Access to the human genome makes it straightforward to identify all genes within a given interval (go to one of the two websites mentioned above) and the ease of DNA sequencing (many companies make this service available) makes it possible to screen candidates by a brute force approach: enough is known about sequence codes to recognize a mutation that will disrupt gene expression, and this can be experimentally verified by looking at the production and structure of mRNA.
is correct. If the likelihood is acceptably high, then the next task is to reduce the genetic interval as much as possible and to identify a causative mutation. Access to the human genome makes it straightforward to identify all genes within a given interval (go to one of the two websites mentioned above) and the ease of DNA sequencing (many companies make this service available) makes it possible to screen candidates by a brute force approach: enough is known about sequence codes to recognize a mutation that will disrupt gene expression, and this can be experimentally verified by looking at the production and structure of mRNA.
A good example of the success of gene mapping in pedigrees is the identification of a mutation in the FOXP2 gene in a family that has a language disorder. The phenotype is complex, including both verbal and non-verbal cognitive impairments. However, the inheritance pattern is straightforward: it fits a model in which a single mutation in one copy of the gene is sufficient to cause disease. By using markers from across the genome, a region on chromosome 7 was identified that co-segregated with the defect and a mutation in a transcription factor, FOXP2, subsequently identified.(27,28)
Pedigree-based linkage works well when the disease follows Mendelian laws of segregation and has led to the identification of many genes involved in intellectual disability and dementia. However, pedigree-based methods for disorders where the genetics does not fit a simple Mendelian pattern have been much less successful. The methods (described in Chapter 2.4.1) typically ask whether affected siblings in a family (usually just a pair) share the same allele at a locus: the more often sharing is observed, the closer the markers locus is inferred to be to the disease gene. The relative failure of the affected sibling pair strategy is almost certainly because of the small contribution that each locus contributes to disease susceptibility.
Neil Risch and Kathleen Merikangas pointed out in 1996 that if the genetic effect size attributable to a single locus is small (that is to say it would increase the risk of developing the disease less than two-fold) then the number of families required using a pedigree method would be impractically large.(29) However, a direct test of association between genetic marker and disease gene had much greater power. Simply by genotyping a marker and determining whether the distribution of genotypes was significantly different between a set of unrelated cases and unrelated controls, small effects could be detected with relatively small sample sizes. The drawback was the need to test variants in every gene, possibly requiring researchers to genotype a million individual markers. Surprisingly, Risch and Merikangas showed that the objection to mounting such a study was not statistical, but technological.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


