Chapter 8 Motor neurone disease
Definition
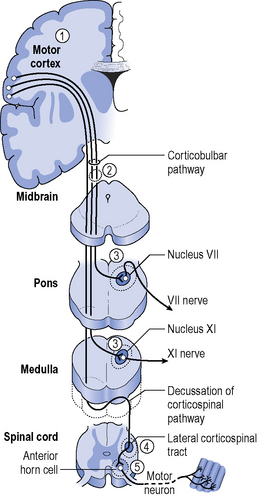
Motor neurone disease (MND) is a degenerative neurological condition characterized by damage at three main sites through the nervous system. Degeneration of anterior horn cells of the spinal cord causes lower motor neurone symptoms. Progressive damage to the corticospinal tracts causes upper motor neurone signs and some motor nuclei in the brainstem are affected causing bulbar palsy (see Figure 8.1).
Aetiology and epidemiology
Although there is no official register of MND patients in the UK, the incidence is thought to be 2 per 100 000 and the prevalence, 7 per 100 000 (Borasio & Miller, 2001). This equates to approximately 5000 people affected in the UK at any one time. Occurrence is slightly higher in men and onset is most common between the ages of 50 and 70 years. Survival and prognosis are discussed later in the chapter (see section on ‘Progression and Prognosis’).
Anatomy and pathophysiology
Involvement of the anterior horn cells and cranial nuclei typically leads to lower motor weakness, wasting and involuntary flickering of muscle fibres, which is known as fasciculations. This atrophy of denervated muscle fibres leads to the use of the term ‘Amyotrophy’(Charcot & Joffroy, 1869).
The exact triggers and pathological processes causing neuronal degeneration remain unknown but current theories suggest it is due to an interaction of multiple factors including genetic predisposition, autoimmune responses, glutamate excitotoxicity and abnormal muscle protein arrangements (Rocha et al., 2005).
Clinical presentation
MND describes a group of disorders where clinical presentation varies and each syndrome or variant is classified depending on the area of the central nervous system involved and the resulting signs with symptoms affecting speech, swallowing, limbs and respiratory muscles (see Figure 8.1):
More recently MND has been found to affect more than just motor neurons and individuals may demonstrate cognitive changes ranging from impaired higher level executive processing in up to one-third to fronto-temporal dementia in approximately 5% (Strong, 2001).
Classical amyotrophic lateral sclerosis
ALS makes up approximately two-thirds of the MND population and is more common in men, with a male:female ratio of 3:2 (Kato et al., 2003). There is involvement of both upper and lower motor neurons in ALS resulting in a mixed picture of upper and lower motor signs in limbs, trunk, respiratory muscles and affecting bulbar functions. Typically there is sparing of certain areas until the later stages of the disease (i.e. Cranial nerves III, IV, VIand spinal nerve S1-S3). This explains the preservation of ocular movement and bladder and bowel function (David, 2002).
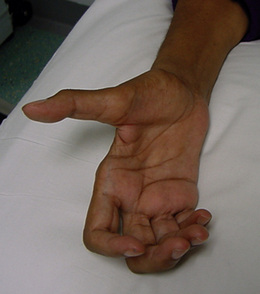
Presentation at diagnosis varies but early signs often include progressive limb weakness, reduced dexterity and wasting of the hands (see Figure 8.2) and approximately two-thirds of those with ALS initially present with symptoms within the limbs. People may also complain of cramps or general fatigue and observe muscle fasciculations. Those with additional bulbar involvement present with wasting of the tongue, muscles of articulation and swallowing, leading to dysarthria and dysphagia. The presence of hyperreflexia in wasted limbs or brisk jaw jerk would indicate the upper motor neuron damage (see Figure 8.2).
Bulbar onset amyotrophic lateral sclerosis
One-third of patients with ALS present with progressive bulbar palsy (Ferguson & Elman, 2007), which affects the action of cranial nerves IX to XII causing dysarthria and dysphagia. When the cranial nerves are affected, the tongue has reduced mobility, is atrophied and fasciculates. Swallowing is often impaired. If the corticobulbar tract is affected, pseudobulbar weakness occurs and the tongue can become spastic leading to dysarthria. Emotional lability may also occur. Bulbar onset ALS is more common in older females with a male:female ratio 2:3 (Mandrioli et al., 2006).
Although initial symptoms are bulbar, the disease usually progresses to include arm, leg and respiratory muscles. The majority of those with ALS have limb or bulbar onset but a small percentage (approximately 5%) may present primarily with respiratory weakness and minimal limb or bulbar involvement (Chen et al., 1996). These patients present with type 2 respiratory failure or signs of nighttime hypoventilation (respiratory issues are discussed in detail later in the chapter).
Overall the above two subsets comprise approximately 90% of the total MND population (Rocha et al., 2005). Typically life expectancy is between 3 and 5 years from symptom onset with an average of 36 months (Haverkamp et al., 1995).
Progressive muscular atrophy/flail limb variant
Syndromes also exist where only one region is involved, e.g. anterior horn cell degeneration in the cervical region leading to flail arm syndrome, or lumbosacral region causing flail leg syndrome. If symptoms are limited to one region the prognosis is significantly better than it is for ALS or PMA where both upper and lower limbs are affected (Talman et al., 2008; Wijesekera et al., 2009).
Primary lateral sclerosis
PLS is a relatively rare condition (affecting 1–3% of those with MND) and presents with upper motor neurone involvement only. Diagnosis is often delayed to exclude potential other causes, but clinically those with PLS have only upper motor neurone signs and normal electromyography of limbs and bulbar muscles (Gordon et al., 2006). The condition can progress to ALS, but if no LMN signs are seen after 4 years prognosis is good with life expectancy comparable to normal, although this is rare.
Diagnosis
Nerve conduction studies (NCS) and electromyography (EMG) help to confirm motor neurone degeneration (Schwartz & Swash, 1995). The motor nerve conduction speed is not affected until advanced stages but the motor action potentials may have reduced amplitude consistent with reduced number of motor axons. Sensory nerve action potentials are usually normal. EMG can identify LMN involvement or denervation prior to clinical signs of wasting and weakness. Multiple areas of denervation are consistent with ALS. If limited to just one nerve root or territory it is more likely to be radiculopathy or a mononeuropathy (Ferguson & Elman, 2007).
At present the diagnosis of ALS is aided by the revised El Escorial criteria where the combination of upper and lower motor neuron signs in the different body regions help guide certainty in diagnosis (Brooks, 2000), although this is not definitive.
Progression and prognosis
To evaluate disease progression it is necessary to establish a baseline using objective measures. Suitable to clinic and research settings the Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS) is a subjective questionnaire regarding functional abilities including speech and swallowing, mobility, personal care and respiratory support (ALS CNTF, 1996). However, a revised version ALSFRS-R (Cederbaum et al., 1999) with an expanded section on respiration has proved more sensitive, better able to predict survival and is now more commonly used. Other outcome measures are discussed relevant to symptoms later in the chapter.
In general, there are several factors indicative of better prognosis: younger age at onset (<65 years), limb as opposed to bulbar onset of symptoms and a longer period between onset of symptoms to diagnosis of MND suggesting a less aggressive disease process (Mandrioli et al., 2006).
At time of writing Riluzole is the only disease-modifying drug available in the UK. A Cochrane review concluded that Riluzole provided modest benefits and increased survival by 2–3 months on average (Miller et al., 2007). Riluzole is a glutamate antagonist thought to inhibit the influx of calcium into the cells, which triggers a series of events that results in neuronal cell death (Lacomblez et al., 1996; Rowland & Shneider, 2001).
Although MND is a progressive, incurable disease it is important to note that quality of life does not directly correspond to level of impairment or disease progression and it does not necessarily decline over time (Simmons, 2005). This further enforces the key role of coordinated support and individualized therapy at all stages of the disease to optimize quality of life.
Multidisciplinary approach
Optimal management of patients with MND requires a team approach, with early referral for clinical assessment and prompt intervention. Multidisciplinary management has been associated with improved quality of life (Van den Berg, 2005) and potentially increased survival time (Traynor et al., 2003). Patients with MND are often seen by several different teams, i.e. community teams, hospice care teams, wheelchair services and social services. Good communication is essential to provide appropriate care for this client group. Clear, patient centred goals are the key to this communication process. The patient must always be included in goal setting and the needs of the carers must also be considered.
Role of the physiotherapist
Symptoms and their management
The motor system
Weakness
Damage to the anterior horn cells leads to a typical lower motor neurone presentation of weakness, flaccidity and atrophy. Suspicion of MND as a potential diagnosis often arises where the classical atrophy of the thenar eminence and dorsal interossei are noticed. In addition to theweakness arising directly from the pathological process of the disease, the MND patient may also face disuse atrophy arising from inactivity and the loss of muscle power that accompanies aging in any individual. Physical inactivity is linked to a decrease in the number of available satellite cells within a muscle and, therefore, a decline in the muscle’s ability to regenerate or recover from damage (Ambrosio et al., 2009).
The role of exercise
Exercise remains a debatable aspect of MND therapy despite an increasing raft of research to underpin its use (Dal Bello-Haas et al., 2008). The fear that it is ineffectual and may even cause further tissue damage still leads therapists to be cautious in its use. However, the benefits of exercise for people affected by MND are not dissimilar to those experienced by the rest of the population (see Ch. 18 for review) and it should feature in the treatment programme of most patients (Dal Bello-Haas et al., 2007). Exercise programmes have been shown to improve ALSFRS scores and to slow decline in physical function (Drory et al., 2001).
Damage to both the upper and lower motor neurones is seen in MND. A summary of the changes in motor neurone activation from each type of damage is presented in Table 8.1. When the motor neurones are damaged by the disease process, the muscle tissue that they innervate can no longer be activated and, therefore, atrophies. These motor units are permanently damaged and cannot be changed by exercise (Peruzzi & Potts, 1996). Other parts of the muscle, however, may have an intact motor neurone and still be functioning. As the patient becomes less mobile and less active due to, for example, fatigue, these otherwise healthy fibres may start to show signs of disuse atrophy. These disused fibres will respond to exercise and may allow the patient to develop a small reserve of healthy, usable muscle. Other factors may affect the muscles of a patient with MND, including increased likelihood of fatigue, impaired respiratory function (resulting in poor delivery of oxygen to the tissues), impaired nutritional intake and hypertonicity. These confounding factors, as well as poor recruitment and retention of subjects in studies amongst this patient group, make clinical research into the effects of exercise very difficult to conduct.
Table 8.1 Respiratory problems
| Respiratory Signs and Symptoms | Cause |
|---|---|
| Dyspnoea at rest | Inspiratory (diaphragm and accessory muscles) weakness |
| Orthopnoea and/or paradoxical abdominal movement | Diaphragm weakness |
| Disturbed sleep, daytime sleepiness | Inspiratory muscle weakness→ Hypoventilation Reduced restful sleep as unable to rely solely on diaphragm |
| Morning headaches and/or altered cognition/confusion | Hypoventilation → carbon dioxide retention |
| Reduced cough | Expiratory (abdominal and accessory muscle) weakness |
| Reduced vocal volume and/or few words per breath | Expiratory muscle weakness |
| Reduced chest wall movement and excessive use of accessory muscles | Inspiratory muscle weakness |
| Paradoxical breathing: Supine-upper abdominals move inwards on inspiration Sitting-intercostals move inwards on inspiration | Negative intrathoracic pressure pulls weak muscles inwards: Diaphragm weakness noted in supine Intercostal weakness noted in sitting |
| Retained secretions | Reduced lung volumes due to inspiratory muscle weakness Reduced cough effort due to expiratory muscle weakness Reduced bulbar function (poor cough trigger and/or poor airway protection- see Bulbar symptoms) |
The primary effect of exercise training is to improve the neural control of the muscle, which will allow more effective recruitment and, therefore, stronger contractions (Sanjak et al., 1987). Since it is only the disused muscle tissue and not the diseased parts that can be strengthened in MND patients, the best results are seen in the least affected muscles.
Studies in other neuromuscular conditions and in mouse models of ALS show that exercise at a submaximal level can be safe and effective (see Ch. 18; Aitkens et al., 1993; Kilmer et al., 1994; Kirkinezos et al., 2003). High-resistance work is thought to be unnecessary and can be damaging to the muscles, particularly with eccentric (muscle-lengthening) contractions, which can cause delayed-onset muscle soreness (Lieber & Friden, 1999). Normal muscle recovers from such damage but repair in diseased muscle may not be possible (Ambrosio et al., 2009). Although eccentric contractions are difficult to avoid within an exercise programme, consideration should be given to how eccentric activity is used to avoid unnecessary strain.
Muscles in patients with MND will fatigue more rapidly than those in healthy individuals. It has not been established whether this is due to impaired central control (Kent-Braun & Miller, 2000) or to impaired activation at a muscular level (Sharma et al., 1995). For this reason, short but frequent exercise sessions may be preferable to prolonged activity.
Recommendations for exercise
Hypertonicity
Hypertonia, a feature of upper motor neurone syndrome, is defined as an increase in resistance to passive stretch and has both a neural (spasticity) and non-neural component (inherent viscoelastic properties of the muscle) which provides resistance to movement and contributes to muscle tone (see Ch. 14). The result of tonal changes can lead to inappropriate movement, discomfort, decreased mobility, reduced function and difficulty with positioning. Spasticity is managed in the same way as for other neurological patients (e.g. stroke, brain injury and multiple sclerosis) and there needs to be a multidisciplinary approach to treatment.
Medical management includes the use of oral medication to reduce tone, such as baclofen, diazepam, dantrolene and tizanadine (Ashworth et al., 2006). Careful monitoring of the effects of medication is needed, as excessive weakness caused by the medication can lead to flaccidity and a reduction in the patient’s functional ability. Intramuscular botulinum toxin injection can be used to reduce focal spasticity (Richardson & Thompson, 1999). The drug weakens the muscle by inhibiting the release of acetylcholine at the neuromuscular junction, preventing muscle contraction. Some patients in the United States have received intrathecal baclofen pumps and achieved a decrease in tone and pain reduction as a result (McLelland et al., 2008). This can be considered but is rarely done in the UK.
Tissue changes
Optimal tissue length is essential if muscle activation and the resultant function are to be maximized. Due to the weakness that results from MND, muscle imbalance causes changes in muscle length. Muscle shortening is especially likely where upper motor neurone involvement leads to increased muscle tone. Therefore, it is imperative that steps are taken early on to prevent changes in tissue length. These may include preventive splinting, stretching programmes and active exercise, positioning, appropriate seating and antispasmodic medication (see other sections of this chapter and Ch. 14).
Physiotherapy management of hypertonicity and resultant tissue changes
 A home exercise programme can be devised for the patient, incorporating stretches to maintain muscle length. As the patient becomes less active, passive stretches can be taught to the carer. Although there is little substantial evidence to support the use of this type of stretching in order to maintain tissue length or influence tone, it may be beneficial in terms of mobilizing joints, providing sensory input and relieving discomfort. Prolonged stretch is required in order to influence muscle length. Splinting may also be used as a means of imposing this sustained stretch (Edwards & Charlton, 2002). Guidelines have been prepared by the Association of Chartered Physiotherapists Interested in Neurology (ACPIN) and these provide information regarding the assessment, procedure, risk factors and protocols for casting. Guidelines for casting patients with complex neurological conditions have also been produced (Young & Nicklin, 2000). Adequate training in splinting techniques is essential.
A home exercise programme can be devised for the patient, incorporating stretches to maintain muscle length. As the patient becomes less active, passive stretches can be taught to the carer. Although there is little substantial evidence to support the use of this type of stretching in order to maintain tissue length or influence tone, it may be beneficial in terms of mobilizing joints, providing sensory input and relieving discomfort. Prolonged stretch is required in order to influence muscle length. Splinting may also be used as a means of imposing this sustained stretch (Edwards & Charlton, 2002). Guidelines have been prepared by the Association of Chartered Physiotherapists Interested in Neurology (ACPIN) and these provide information regarding the assessment, procedure, risk factors and protocols for casting. Guidelines for casting patients with complex neurological conditions have also been produced (Young & Nicklin, 2000). Adequate training in splinting techniques is essential. The theory behind weight-bearing activities is to maintain joint range and stimulate antigravity muscle activity (Brown, 1994). Standing exercises can be performed independently by the patient, with assistance, or with the use of specialized equipment such as a tilt table or Oswestry standing frame.
The theory behind weight-bearing activities is to maintain joint range and stimulate antigravity muscle activity (Brown, 1994). Standing exercises can be performed independently by the patient, with assistance, or with the use of specialized equipment such as a tilt table or Oswestry standing frame. Different postures and positions have an influence on tone and movement. If a patient is uncomfortable, then this may lead to an increase in muscle tone. Therefore, correct positioning over a 24-hour period is vital. Liaison with the local wheelchair team is important to provide the patient with a suitable wheelchair (see ‘Positioning and seating’ section).
Different postures and positions have an influence on tone and movement. If a patient is uncomfortable, then this may lead to an increase in muscle tone. Therefore, correct positioning over a 24-hour period is vital. Liaison with the local wheelchair team is important to provide the patient with a suitable wheelchair (see ‘Positioning and seating’ section). If a task is very effortful for a patient, then this may increase muscle tone and lead to difficulties completing the task. Therefore, activity modification is advised. Decreasing the effort of the task by either activity modification or the use of aids and adaptations may assist with reducing tone.
If a task is very effortful for a patient, then this may increase muscle tone and lead to difficulties completing the task. Therefore, activity modification is advised. Decreasing the effort of the task by either activity modification or the use of aids and adaptations may assist with reducing tone. A noxious stimulus such as constipation, skin breakdown or an in-growing toenail may lead to an increase in tone. Poor positioning or an ill-fitting orthotic appliance may also increase tone unnecessarily. Therefore, it is important to identify any cause and make adjustments or withdraw the cause when managing patients with hypertonia.
A noxious stimulus such as constipation, skin breakdown or an in-growing toenail may lead to an increase in tone. Poor positioning or an ill-fitting orthotic appliance may also increase tone unnecessarily. Therefore, it is important to identify any cause and make adjustments or withdraw the cause when managing patients with hypertonia.Fatigue
Patients with MND frequently experience generalized fatigue, which can be one of the most disabling features of the condition. However, with good advice, they can learn how to minimize the effect of this problem. Activity should be encouraged but not so as to exhaust the patient (see ‘The role of exercise”, above).
Cramps
People with MND may experience painful cramps, often at night. These can be alleviated with stretching and massage. In severe cases, Quinine Sulphate can be given to prevent the cramping (Miller et al., 2005). Cramps will often be worse in patients who display hypertonicity and good management of the tone problems will help to minimize the other muscle-based problems.
Resultant disabilities
Any combination of the above impairments may lead to significant disability. These include:
Immobility
Transfers may become difficult and advice from the physiotherapist on safe and effective technique may help to maintain independence in this activity. Many devices are commercially available to improve transfers, such as sliding boards, transfer belts and hoists. Transfer belts placed around the lower thorax/pelvis can be particularly useful where the patient has flail arms or painful shoulders, allowing the carer to have a firm hold on the patient without the risk of trauma to the limbs. However, this requires the patient to be able to maintain an upright posture and, therefore, may not be appropriate in the late stages of the disease. Consideration of comfort, type of transfer and environment should be made when selecting a hoist. Manual handling legislation must be part of the reasoning process when selecting transfer techniques (RCN, 1999).
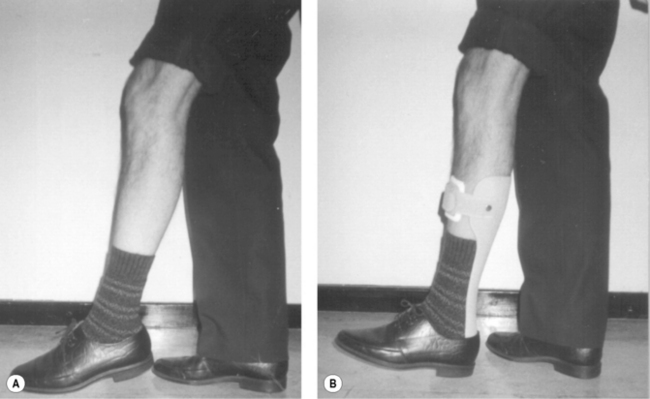
Orthoses may also be necessary to maximize mobility. Lively or rigid splints and light weight orthoses can be of use, but careful assessment of their value must be made frequently. An ankle-foot orthosis is often provided for loss of active dorsiflexion (Figures 8.3A & B) and a ‘foot–up’ orthosis (Figure 8.4) is often beneficial in the early stages where mild or fatiguing footdrop presents.

Due to the progressive nature of MND, early referral for wheelchair provision is essential. Patients’ local wheelchair service should be able to provide a chair that meets their mobility and postural needs, but if the patient desires something beyond this remit, there is a large selection of wheelchairs available commercially which have other features. If a wheelchair is being bought privately, it is worthwhile seeking advice from the wheelchair service to ensure it fully meets the patient’s requirements and to investigate voucher scheme funding. The wheelchair may be manually propelled by the patient or attendant, or powered for indoor and/or outdoor use. Scooters are not currently provided by the National Health Service, but may provide another mobility option. Consideration must be given to postural requirements, function and pressure care when providing a wheelchair and seating system (Trail et al., 2001).
Loss of head control
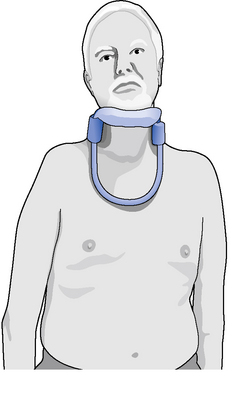
There are several specially designed collars available that may offer a solution to these problems (Figure 8.5). These include two specifically designed rigid head supports, the Headmaster and the MNDA (Mary Marlborough Lodge) collar. Both provide a flexible platform for the chin that allows a small amount of anteroposterior movement for speaking and chewing, but prevents the head falling forwards, and they are open at the front to avoid throat compression.





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


