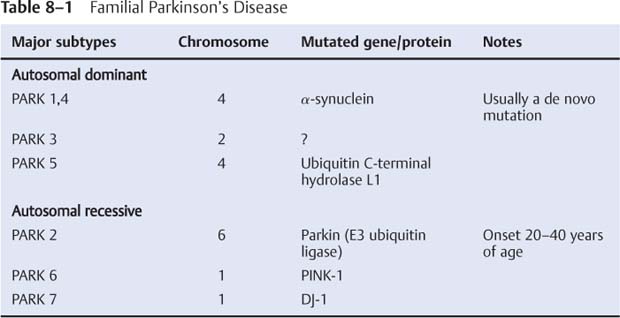
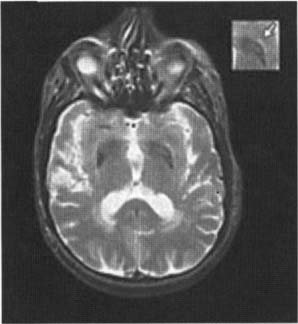
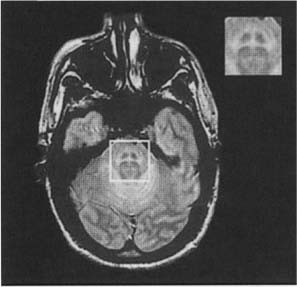
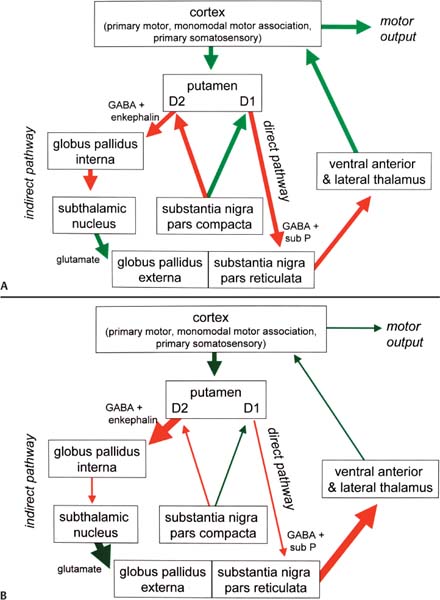
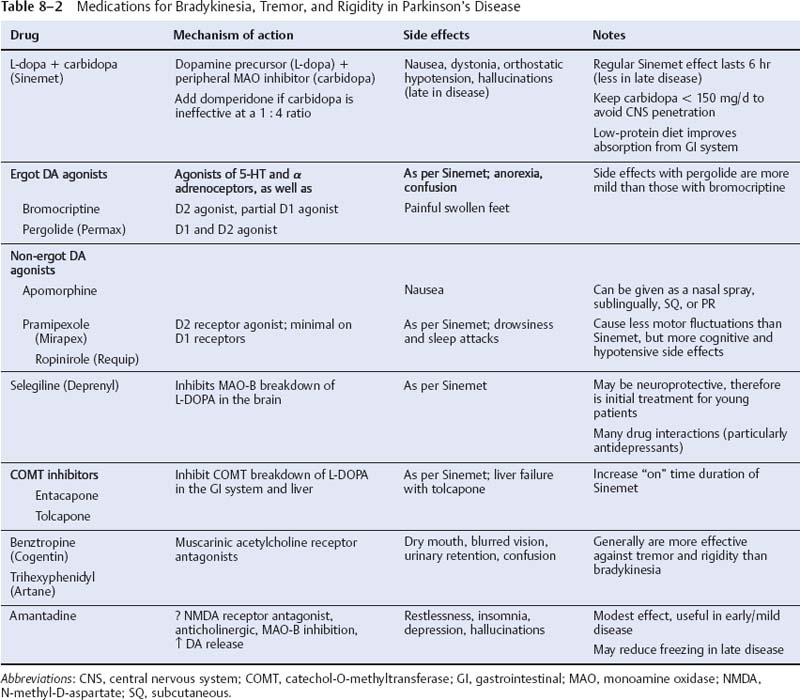
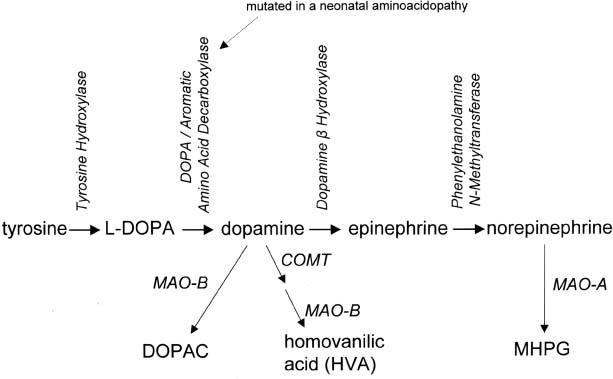
8 Note: Significant diseases are indicated in bold and syndromes in italics. 1. Pathophysiology: increased inhibition of the ventral anterior and lateral thalamic output to the motor cortex caused by loss of dopaminergic input to the striatum (Fig. 8–1) a. D2 receptors in the striatum are expressed on aspiny and spiny neurons (see p. 11); activation of the D2 receptor causes acetylcholine release onto muscarinic receptors that inhibits glutamatergic input to the striatum from the cortex and the GABAergic neurotransmission within the basal ganglia (i.e., D2 receptor activation inhibits most of the basal ganglia circuit) b. histology: degeneration of the dopaminergic substantia nigra pars compacta neurons and, to a lesser extent, the basal forebrain (e.g., nucleus basalis of Meynert), the hypothalamus, and brainstem pigmented neurons (locus coeruleus, dorsal vagal motor nucleus) probably due to dysfunction in the ubiquitin–proteosome system i. neuron loss is pronounced in lateroventral part of pars compacta, and is usually asymmetric; symptoms appear typically when 80% of substantia nigra pars compacta neurons are lost ii. surviving neurons often exhibit eosinophilic inclusions surrounded by a halo that contain α-synuclein and ubiquitin {Lewy body (see p. 162)} (Box 8.1) (1) α-synuclein is a presynaptic protein thought to be involved in dopamine release and/or synaptic plasticity; in Parkinson’s disease it becomes insoluble and aggregates into fibrils that attract ubiquitin and neurofilaments, thereby forming Lewy bodies c. genetics: the majority of cases are sporadic, but several inherited forms exist (Table 8–1) i. parkin: acts to covalently link proteins targeted for destruction to ubiquitin, which then can be recognized by proteosomes; mutations cause accumulation of intracellular proteins that form Alzheimer’s-like neurofibrillary tangles instead of Lewy bodies (1) accounts for the majority of early-onset Parkinson’s disease ii. ubiquitin C-terminal hydrolase L1 acts to release ubiquitin from its conjugated protein after delivery of the conjugated protein to the proteosome iii. PINK1: a mitochondrial protein kinase that regulates the electron transport chain 2. Epidemiology: exhibits increased incidence according to geographic (industrialized countries > nonindustrialized countries) and racial factors a. risk factors for sporadic Parkinson’s disease include drinking well water, rural residence, farming (possibly related to pesticide exposure in industrialized countries), and employment in wood pulp mills or steel industries; risk may be reduced by cigarette smoking and caffeine use Figure 8–1 (A) normal basal ganglia function. (B) Basal ganglia function in Parkinson’s disease. a. initially presents as a unilateral resting tremor, bradykinesia, or limb stiffness; progressive rigidity and bradykinesia eventually will become bilateral and will involve freezing episodes and dysphagia i. progression involves hypophonia, dysarthria, dysphagia, and an expressionless face ii. symptoms are slowly progressive, presenting asymmetrically or unilaterally iii. symptoms most commonly begin in the upper extremities, but occasionally may initiate in the lower extremities or jaw (particularly with tremor) b. depression (50%) c. dementia (30%): half of demented Parkinson’s disease patients have no other pathological cause, but 35% exhibit Alzheimer’s-like histological changes d. restless legs syndrome; akathisia; REM sleep behavioral disorder (see pp. 169–170) e. autonomic symptoms (urinary urgency, constipation {intestinal pseudo-obstruction}, sexual dysfunction) and oculomotor abnormalities are mild and develop only late in the disease course i. 20% of patients develop significant orthostatic hypotension often due to medication side effects, but this can represent the misdiagnosis of a multisystem atrophy disorder f. dementia develops in 30% of patients with advanced disease 4. Treatment (Table 8–2) i. management of autonomic dysfunction (1) orthostatic hypotension (a) nonpharmacological treatments: graded position changes; avoid Valsalva maneuvers; squatting or flexing of the lower extremity musculature prior to standing; increase fluid and salt intake; lower extremity compressive garments (b) pharmacological treatments (i) fludrocortisone (ii) midodrine, ephedrine, pseudoephedrine, methylphenidate (iii) desmopressin acetate (ddAVP); erythropoietin; caffeine (2) bladder hyperactivity: reduce drinking in the evening; anti-cholinergic medications (oxybutynin, propantheline) (3) constipation: dietary modification and stool softeners; discontinuing anticholinergic medications ii. management of side effects of medical treatment (1) “off”/freezing phenomenon (a) combine a dopamine agonist with Sinemet (b) increase the frequency of combined medications (c) substitute or add a sustained-release preparations of Sinemet (2) dyskinesia/dystonia (a) occurring with the peak in drug levels (“on” dyskinesia/dystonia) (i) lower the dose of dopamine agonists or Sinemet (ii) substitute sustained-release preparations of Sinemet for shorter-acting medications (iii) add a catechol-O-methyltransferase (COMT) antagonist (iv) consider neurosurgical intervention (b) occurring a minimal drug levels (“off” dyskinesia/dystonia) (i) evening “off” dyskinesia: add sustained-release Sinemet in the evening (ii) between dose “off” dyskinesia 1. add dopamine agonist or amantadine 2. increase the frequency of medication administration 3. consider neurosurgical intervention (3) treatment-induced hallucinations (a) identify and discontinue the offending medication in order of likely causation: anticholinergics > amantadine > selegiline > dopamine agonists > Sinemet (b) lower the dose of dopamine agonists or Sinemet (c) add a low-dose atypical antipsychotic medications (clozapine, quetiapine) (Box 8.2) b. surgical treatment: best response to medication postoperatively is equal to the patient’s best response to medication preoperatively, and the chief improvement is in terms of reduced fluctuations and dyskinesias i. lesion procedures: usually done with stereotactic and intraoperative electrophysiological confirmation of location, but can be done by means of radiosurgery (e.g., gamma knife) (1) thalamotomy: targets the ventral intermediate nucleus (a) improves only tremor, therefore not often used in Parkinson’s disease (b) not effective against bradykinesia, and may exacerbate hypophonia and gait abnormalities; avoid bilateral procedures due to cognitive impairment (2) pallidotomy: targets the posterolateral region of the internal globus pallidus to avoid impairment of memory and cognitive function (a) effective against all major Parkinson’s disease symptoms and dyskinesias (b) limited to a unilateral procedure due to high incidence of cognitive impairment with bilateral procedures ii. deep brain stimulator procedures: over stimulation may produce dyskinesias (1) thalamic stimulation: effectiveness as per thalamotomy, but fewer side effects (2) globus pallidus stimulation: effectiveness as per pallidotomy, but fewer side effects; bilateral stimulation has pronounced benefits for gait abnormalities (3) subthalamic stimulation: most effective surgical procedure for bradykinesia, and is effective for all other major Parkinson’s disease symptoms; bilateral stimulation has pronounced benefits for gait abnormalities The tau-opathies—Alzheimer’s disease, frontotemporal lobar degeneration/Pick’s disease; cortical-basal ganglionic degeneration; progressive supranuclear palsy 1. Pathophysiology: Gliosis and neuron loss predominantly in the frontal cortex, globus pallidus, substantia nigra, and mesencephalic nuclei a. histology: affected areas exhibit Alzheimer’s disease-like neurofibrillary tangles (with hyperphosphorylated tau protein) and neuropil threads i. D2 receptors in the basal ganglia are reduced, unlike Parkinson’s disease where they are increased; this likely reflects the loss of cholinergic striatal interneurons in PSP (see p. 11) b. genetics i. 90% of sporadic cases are associated with homozygous H1 alleles of the tau gene, although 60% of normal people also are homozygous for the H1 allele ii. families with autosomal dominant inheritance patterns have been described 2. Symptoms: develop between 60–70 years of age a. axial rigidity > limb rigidity, leading to an abnormal gait and falls early in the disease (particularly backward falls, unlike Parkinson’s disease patients who fall forward) b. cortical-type dysarthria; hypophonia c. reduced voluntary saccades, particularly a vertical gaze palsy involving downward > upward gaze; voluntary smooth pursuit movements become impaired thereafter i. involuntary (vestibulo-ocular) eye movements are preserved until late in the disease 3. Diagnostic testing: loss of saccadic fast-phase on optokinetic testing may distinguish early PSP from Parkinson’s disease 4. Treatment: none specific 5. Prognosis: usually fatal within 6 years 1. Pathophysiology: Gliosis with neuronal loss and vacuolation in cortical layers III–V (like frontotemporal lobar degeneration/Pick’s disease), the basal ganglia, and the substantia nigra; depigmentation of the substantia nigra also occurs a. surviving neurons exhibit accumulations of hyperphosphorylated tau b. genetics: associated with the H1 allele of the tau gene, as per PSP 2. Symptoms: develop > 60 years of age; symptoms occur in a random order and may relapse and remit, but they all are concentrated initially in one limb a. subcortical symptoms: Parkinsonism; postural and/or action tremor; limb dystonia; reflex myoclonus b. cortical symptoms: dementia similar to frontotemporal lobar degeneration (see p. 160); apraxia (e.g., constructional dyspraxia); cortical sensory loss; weakness i. Gerstmann’s syndrome and alien-hand syndrome is a rare complication of corticobasal ganglionic degeneration 3. Diagnostic testing: MRI may reveal regional degeneration in frontal and parietal cortex 4. Treatment: none specific 5. Prognosis: symptoms remain focally in one limb for several years, but eventually progress to involve other limbs; severe dysphagia eventually develops 1. General histology: exhibits multiple types of inclusion bodies to varying degrees; the most typical for MSAs are a. an argentophilic cytoplasmic inclusion in neurons and glia that resemble neuropil threads but that contain only ubiquitin and no tau protein b. a flame-shaped glial cytoplasmic inclusion similar to the neurofibrillary tangles of Alzheimer’s disease but that do not contain tau protein 2. The old classification scheme a. olivopontocerebellar degeneration (OPCD)—idiopathic degeneration of the inferior olivary nuclei, ventral pontine nuclei, and cerebellar cortex with relative preservation of the substantia nigra and striatum b. striatonigral degeneration—idiopathic degeneration of the putamen and substantia nigra with relative preservation of the globus pallidus, caudate, and subthalamic nucleus c. Shy-Drager syndrome—idiopathic degeneration of the intermediolateral cell column in thoracic and lumbar spinal cord, the dorsal motor nucleus of the vagus, and in the sympathetic chain ganglia 3. The new classification scheme: The Shy-Drager syndrome has been eliminated because autonomic dysfunction is present in all types of multiple-system atrophy a. multiple system atrophy with predominant parkinsonian features (MSA-P)/striatonigral degeneration form of MSA i. histology: the pattern of degeneration is as for striatonigral degeneration Shy-Drager syndrome (1) despite pronounced Parkinsonian features, does not have Lewy bodies, neurofibrillary tangles, or even many of the MSA inclusion bodies in the surviving neurons of substantia nigra ii. epidemiology: sporadic occurrence only iii. symptoms: bradykinesia, rigidity, autonomic dysfunction; less commonly involves myoclonus, neck anteroflexion, or dysarthria (1) autonomic dysfunction usually predates parkinsonian features (2) hyperpigmentation of the extremities iv. diagnostic testing: late in the disease, MRI demonstrates (1) T2 hypointensity in the globus pallidus with a lateral linear hyperintensity, and putamen atrophy (Fig. 8–2) (2) loss of pontine fibers {hot-cross bun sign} (Fig. 8–3) v. treatment: dopaminergic agents usually have little effect against the parkinsonism because D2 receptors are lost in the striatum; management of autonomic dysfunction as per Parkinson’s disease b. multiple system atrophy with predominant cerebellar features (MSA-C)/OPCD form of MSA i. histology: pattern of degeneration is as for olivopontocerebellar degeneration Shy-Drager syndrome ii. epidemiology: peak incidence at 50–60 years of age; usually sporadic, but may have autosomal dominant or recessive patterns of inheritance (usually such cases are classified under spinocerebellar ataxias) iii. symptoms: ataxia in the lower extremities > upper extremities; cerebellar-type dysarthria; autonomic dysfunction (Box 8.4) (1) subtypes of MSA-C include (a) parkinsonian symptoms (50%) (b) retinal degeneration (c) spastic paraplegia and areflexia (d) dementia, ophthalmoplegia, and extrapyramidal signs iv. diagnostic testing: late in the disease, neuroimaging demonstrates atrophy of the brainstem and cerebellum v. treatment: none specific; management of autonomic dysfunction as per Parkinson’s disease Figure 8–2 T2 MRI of MSA-P. (From Kaufmann H, Biaggioni I. Autonomic failure in neurodegenerative disorders. Semin Neurol 2003, 23:357, Fig. 3A. Reprinted by permission.) MSA-C usually occur in a hereditary pattern, and thus are considered spinocerebellar ataxias (see p. 198). 1. Pathophysiology: primary dystonias result from dysfunction the dopaminergic systems (Fig. 8–4) in the of basal ganglia without apparent neuron loss or neurochemical deficit; abnormal function of the motor and sensory cerebral cortex and in lower motor neuron reflexes are likely reactions to the basal ganglia dysfunction Figure 8–3 Hot-cross bun sign of MSA-P. (From Kaufmann H, Biaggioni I. Autonomic failure in neurodegenerative disorders. Semin Neurol 2003, 23:357, Fig. 3C. Reprinted by permission.) a. primary and secondary motor and sensory cerebral cortex exhibit abnormal activation patterns even when dystonic movements are not occurring b. lower motor neuron reflexes are apparently abnormal because the dystonic movements involve unnecessary activation of co-agonist muscles and also antagonist muscles i. even when dystonic movements are not occurring, dystonia patients exhibit reduced skeletal muscle inhibition (e.g., reduced reciprocal inhibition of antagonist muscles during voluntary movement in focal dystonia patients, reduced suppression of the blink reflex in blepharospasm patients) c. focal lesions can cause dystonia when they are located in the contralateral putamen > globus pallidus >> caudate, thalamus, brainstem i. rarely dystonia presents after peripheral nerve injury, likely by altering basal ganglia function in genetically susceptible individuals 2. Causes of dystonia Figure 8–4 Biosynthesis of dopamine, norepinephrine, and epinephrine. a. primary dystonias (Table 8–3) b. secondary dystonias i. neurodegenerative disorder-related dystonias: Parkinson’s disease, multiple-system atrophies, progressive supranuclear palsy, corticobasal ganglionic degeneration, spinocerebellar atrophies (particularly Machado-Joseph’s disease/SCA-3), ataxic telangiectasia, Huntington’s disease, dentatorubropallidoluysian atrophy (DRPLA)
Movement Disorders
I. Parkinson’s Disease (Box 8.1)
II. Progressive Supranuclear Palsy (PSP) (Box 8.3)
Box 8.3
III. Cortical-Basal Ganglionic Degeneration
IV. Multiple System Atrophy (MSA)
Box 8.4
V. Dystonias
Type | Genetics | Notes |
Early-onset dystonia (DYT1) | Mutation in torsin A gene, an ATP-binding protein (AD, 60% penetrance) | Childhood onset Progresses to other limbs Accounts for most primary dystonia |
Dystonia-Parkinsonism/Lubag syndrome | Unknown gene (X-linked) | Segmental or diffuse dystonia Common only in Philippines |
DOPA-responsive dystonia-Parkinsonism | Mutation in GTP cyclohydrolase I gene (AD) (Box 8.5) | Treat with levodopa–carbidopa (Sinemet) |
Segawa syndrome | Mutation in tyrosine hydroxylase gene (AR) (Box 8.6) |








