Nabila A. Dahodwala
Howard I. Hurtig
Several movement disorders increase in incidence and prevalence with old age. However, normal aging is associated with changes (e.g., slowed motor speed) that can resemble common findings of movement disorders. Also, older adults are more likely to take multiple prescription medications and, therefore, are at risk for developing drug-induced movement disorders (81). Thus, the clinician caring for the older adult must distinguish between age-related, normative findings and true movement disorders as well as target therapeutic interventions to the specific needs of the elderly patient.
HYPOKINETIC MOVEMENT DISORDERS: PARKINSONISM
For clinical purposes, movement disorders can be divided into hypokinetic and hyperkinetic disorders. Parkinsonism, the most common of the hypokinetic disorders, is a “syndrome” of slowed movements, rigidity, postural instability, and tremor. Idiopathic parkinsonism or Parkinson’s disease (PD) is the most common parkinsonian syndrome, accounting for approximately 75% of the total number of patients with a parkinsonian diagnosis (78). Of the select group of patients that present to autopsy, idiopathic PD represents about 55% of patients with parkinsonism seen at specialty clinics (40). PD is a progressive, degenerative disease of the brain with distinctive clinical features; a specific histopathology characterized by degeneration of neurons throughout the brain, especially pigmented, dopamine-producing neurons in the substantia nigra (SN) of the midbrain; and a strongly positive response to dopamine replacement therapy with levodopa by most patients. Parkinsonism caused by other degenerative disorders such as progressive supranuclear palsy (PSP), multiple system atrophy (MSA), corticobasal degeneration (CBD), adverse effects of drugs (mainly neuroleptics), stroke, and other rare miscellaneous causes comprise the other 25% of cases.
Parkinsonism is a disorder of aging. PD is uncommon before the age of 40, and incidence and prevalence increase steadily until the ninth or 10th decades (96). Although PD can begin in childhood or early adulthood (juvenile PD), the mean age of symptom onset is about 70 years of age (100).
PATHOGENESIS
The cause of PD is unknown. However, the leading hypothesis of causation holds that the pathogenesis in the majority of cases represents a complex interaction of genetic and environmental factors. In the last decade, several genes and genetic loci have been identified that are both dominantly and recessively inherited with varying degrees of penetrance. Thus far, these genes and their corresponding loci include α-synuclein/PARK1 and 4 (dominant), LRRK2/PARK8 (dominant), parkin/PARK2 (recessive), PINK1/PARK6 (recessive), and DJ-1/PARK7 (recessive). Understanding the normal function of these proteins and how the altered protein structures lead to PD will help to shed light on the pathophysiology of PD in both genetic and nongenetic cases (35).
The pathology of PD, on the other hand, has been well described for almost a century. Patients with PD who are autopsied are usually severely disabled. At postmortem examination in such a patient, the gross brain sliced transversely at the level of the midbrain shows pallor compared to the dark, linear, mustacheshaped stripe that marks the presence of a population of the approximately 400,000 pigmented neurons in the normal SN. A microscopic view of the SN in PD shows marked depletion of these neurons. The severity of the clinical disability in PD is usually proportional to the loss of dopaminergic neurons in the SN. The histopathologic hallmark of the disease is the eosinophilic, intraneuronal, intracytoplasmic inclusion known as the Lewy body (LB), named for Frederick Lewy, the German neuropathologist who first described the inclusion in 1912. LBs can be found in the SN, locus ceruleus, dorsal motor nucleus of the vagus nerve, thalamus, hypothalamus, substantia innominata (nucleus basalis of Meynert), and cerebral cortex, especially in older patients with a long history of PD who develop dementia (1,59).
LBs consist of the accumulation of a protein called alpha-synuclein whose normal function is believed to involve modulation of synaptic vesicle turnover and synaptic plasticity (14). Several hypotheses exist that explain how these protein aggregates develop and how they may then lead to neuronal death. These include exposure to neurotoxins and oxidative stress and genetic mutations that lead to altered protein
folding, polymerization, and degradation (27). There is no single biochemical pathway to date that explains the development of LBs. However, active investigation continues in the field because it provides hope for more targeted and potentially curative therapies.
folding, polymerization, and degradation (27). There is no single biochemical pathway to date that explains the development of LBs. However, active investigation continues in the field because it provides hope for more targeted and potentially curative therapies.
CLINICAL MANIFESTATIONS
The diagnosis of PD is based on a clinical impression of PD because there are no tests currently available to validate what a doctor finds after interviewing and examining a patient. Computerized imaging techniques, such as computed tomography (CT) and magnetic resonance imaging (MRI), usually are normal in the setting of PD or show only nonspecific changes. Computed isotopic imaging, such as positron emission tomography (PET) and single photon emission CT (SPECT) have shown promise in differentiating patients with PD or another parkinsonian syndrome from patients whose illness has a nonparkinsonian cause, but these techniques are not widely available.
The cardinal features of PD—rest tremor, rigidity, and bradykinesia—usually appear in the limbs asymmetrically, often affecting only one side of the body in the early stages of the disease. Most movement disorder specialists believe that a patient must have two of the three cardinal features and a strongly positive response to levodopa before a clinical diagnosis of PD can be considered definite (39). Patients with other parkinsonian syndromes (e.g., PSP, MSA, and CBD) rarely respond as well to levodopa as do patients with PD. Postural instability (tendency to fall) is a relatively late feature of PD, in contrast to the other parkinsonian syndromes, in which it appears relatively early in the course.
Tremor, often the initial symptom, is eventually present in 75% of patients with PD. It has a frequency of 5 to 7 Hz and usually starts on one side in a hand or foot (24). Tremor is typically most prominent when the affected part is at rest, but it can also be evident with sustention and action. As the disease progresses, the tremor may involve both sides as well as the lips and chin. Rigidity (increased tone) or “stiffness” is a common complaint and can be detected when an examiner observes resistance to passive movement in a relaxed limb that occurs throughout the whole range of movement. Increased tone that starts and stops as the limb is moved passively back and forth is called “cogwheeling.” Rigidity is an inconstant finding but can occur in all stages of disease. Bradykinesia refers to slowness of initiating and sustaining movement; it is different from rigidity and can be distinguished from it through a careful examination because rigidity is often not present in a patient who is noticeably slow. Bradykinesia and tremor are the most noticeable and constant physical signs of parkinsonism. Bradykinesia underlies many of the symptoms of PD, including difficulty with dexterity, fine motor movements, and gait.
Gait dysfunction in PD reflects a composite of bradykinesia, rigidity, and impairment of postural righting reflexes. Stride length is shortened, and the feet shuffle. Arm swing is decreased, more so on the more affected side. Posture is stooped, and the arms assume a flexed and adducted position. Turning in the act of walking is done en bloc, without pivoting or twisting the torso and with multiple small steps that describe a wide arc. As the disease progresses, the gait is interrupted by transient freezing of the feet, as though they were glued to the ground, especially on initiating the first step, when entering a narrow space such as a doorway, or when turning at an acute angle.
Loss of balance and falling tend to occur later in the course of PD. Patients who present with postural instability or frequent falls early in the course of illness should be evaluated for other causes of parkinsonism such as PSP or MSA. A modest backward pull on the patient’s shoulders can be used to test postural instability. The patient should be instructed to take only one step back if necessary to prevent going backward. If more than two steps are required to regain balance after a reasonably forceful tug (not too hard!), postural reflexes are impaired. More severely affected patients may fall like solid objects (95), and the examiner must be prepared to catch the patient. Stooped posture and a loss of postural reflexes can lead to an accelerated speed of walking, forward leaning of the patient’s torso, and a tendency to fall, as the patient “chases his or her center of gravity.” This type of accelerated forward locomotion is referred to as a “festinating gait.”
There are many secondary manifestations of PD (Table 22-1), which are classified as “nonmotor” symptoms but are not sufficient by themselves to confirm a diagnosis of the disease. However, some secondary symptoms may be the presenting complaint and may be more debilitating than the primary features of PD.
PD remains a clinical diagnosis, despite recent advances in brain imaging. Computerized isotopic brain imaging (PET and SPECT) and transcranial sonography make it possible to visualize the abnormalities in the basal ganglia caused by PD. The University of Pennsylvania Smell Identification Test (UPSIT) can detect the hyposmia (loss of the sense of smell) that is often seen in the earliest stage of clinical PD. These diagnostic tests are primarily used in research with the goal of detecting early or preclinical PD (92), but PET or SPECT imaging can be useful in the right setting to confirm or refute an uncertain clinical diagnosis.
Table 22-1. Secondary Symptoms of Parkinson’s Disease | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
DIAGNOSING PARKINSON’S DISEASE IN THE ELDERLY
Some signs of parkinsonism can be a part of normal aging. In one study, bradykinesia was present in 37% of normal elderly community residents (81). No increase was noted in tone in normal elderly subjects, but joint stiffness caused by arthritis can limit mobility and be confused with increased tone. Similarly, posture tends to become flexed at the neck and trunk as part of normal aging. Dizziness can lead to gait instability, and stride length is shorter in the elderly (71). In a community sample of older adults, 15% of people 65 to 74 years of age and almost 30% of people 75 to 84 years of age had some evidence of parkinsonism (7). This, however, was also associated with a twofold increased risk of mortality and more likely represents a complex interaction of aging, neurologic disease, medication effects, and vascular disease as the etiology of parkinsonism. The descriptive terms “senile gait” and “old person’s gait” are two of the many default labels used to define the nonspecific but sometimes incapacitating Parkinson-like abnormality of walking by the elderly when no obvious other cause can be found. Rest tremor does not occur as a manifestation of normal aging and is one of the more specific indicators of PD in the elderly. If bradykinesia and postural instability alone were used in the diagnosis of the disease, many normal elderly patients might be misdiagnosed. Therefore, to make a diagnosis of PD, all patients, young or old, should meet the standard criteria of two of the three cardinal features and a good response to levodopa.
OTHER CAUSES OF PARKINSONISM IN THE ELDERLY
Most cases of parkinsonism are ultimately classified as PD, although the differential diagnosis of secondary parkinsonism is broad (Fig. 22-1). Structural lesions of
the basal ganglia (stroke, hydrocephalus, and tumor), drugs, infections, or metabolic disorders can rarely mimic typical PD. In general, these patients do not have a rest tremor and do not respond to levodopa (66).
the basal ganglia (stroke, hydrocephalus, and tumor), drugs, infections, or metabolic disorders can rarely mimic typical PD. In general, these patients do not have a rest tremor and do not respond to levodopa (66).
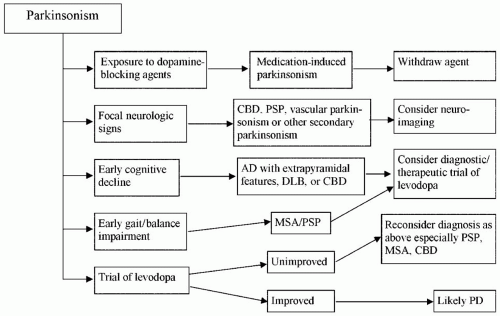 Figure 22-1. Approach to the parkinsonian patient. |
Vascular Parkinsonism
Parkinsonism can occur rarely after a single stroke in the basal ganglia or brainstem or as the cumulative result of several strokes. Patients with vascular parkinsonism tend to have a long history of hypertension and a stepwise progression of symptoms that may correlate with each acute clinical stroke. Evidence for stroke may be noted on examination. Gait may be shuffling or magnetic, a term used synonymously with transient freezing. Patients should have evidence of cerebrovascular disease on CT scan or MRI, although asymptomatic ischemic changes are common in older patients and do not independently make the diagnosis of vascular parkinsonism (43).
Hydrocephalus
Hydrocephalus can cause parkinsonian signs and symptoms. Normal pressure hydrocephalus (NPH) is a syndrome characterized by the triad of gait instability, urinary incontinence, and cognitive dysfunction (55,89). As with vascular parkinsonism, gait in NPH is classically magnetic, but other gait disturbances, especially “gait ignition failure” and “frontal gait” (67), have been described in NPH. Brain imaging (CT or MRI) shows communicating hydrocephalus with ventricles enlarged out of proportion to atrophy of the cerebral cortical sulci. This is an important disorder to consider in any patient with a parkinsonian gait, cognitive impairment, and urinary frequency or incontinence because ventriculoperitoneal shunting may lead to the dramatic reversal of a severe disability. For this reason, every patient with parkinsonism deserves to have a computed imaging study of the brain, either CT or MRI, at some time during the course of illness; the sooner it is performed the better to get the question out of the way.
Drug-Induced Parkinsonism
Drug-induced parkinsonism is a critically important entity because it is one of the few reversible causes of parkinsonism. The most common offenders are the antipsychotics, also known as neuroleptics, which as a class can cause parkinsonism because of their dopamine receptor-blocking properties. These include phenothiazines, butyrophenones, and thioxanthenes (45). The antiemetic prochlorperazine (Compazine) and the gastrointestinal promotility agent metoclopramide (Reglan) are also dopamine receptor blockers that have the potential to induce parkinsonism and other “extrapyramidal” involuntary movements, such as dystonia and tardive dyskinesia (TD) (44). Clozapine (Clozaril), the first “atypical” neuroleptic, was introduced to the American market in 1990 after prior experience in Europe as a major breakthrough for treating schizophrenia because its antipsychotic effect is mediated by nondopaminergic neural circuits. Therefore, it relieves symptoms of psychosis without causing parkinsonism or other extrapyramidal side effects. Several other atypical neuroleptics have followed, including olanzapine (Zyprexa), risperidone (Risperdal), ziprasidone (Geodon), and aripiprazole (Abilify), although each has greater affinity for the dopamine receptor than clozapine and can, therefore, produce varying degrees of extrapyramidal side effects. Quetiapine (Seroquel) is one of the more recently developed atypical neuroleptics with an extrapyramidal side effect profile similar to clozapine. Other drugs with a tendency to induce parkinsonism include the antihypertensives methyldopa, verapamil (38), reserpine, and the antiepileptic valproic acid (Depakote). Reserpine can induce signs of parkinsonism by depleting dopamine storage vesicles presynaptically instead of blocking postsynaptic dopamine receptors. Treatment of parkinsonism in this setting consists of a combination of a high index of suspicion and prudent withdrawal of the offending drug.
Alzheimer’s Disease and Dementia with Lewy Bodies
Neurodegenerative diseases other than PD often have clinical parkinsonian features. Alzheimer disease (AD), the most common cause of dementia, may be accompanied by varying degrees of parkinsonism in up to 50% of patients (13), usually late in the course of the dementia. In AD, the dementia precedes the parkinsonian signs and is disproportionately more severe. Dementia and parkinsonism also co-occur in the pathologic entity diffuse LB disease, also known clinically as dementia with LB (DLB), which is associated pathologically with widespread distribution of LBs throughout the cerebral cortex. The clinical presentation in DLB consists of prominent and early cognitive decline, fluctuations in cognitive function, spontaneous visual hallucinations that have no other cause, and parkinsonism (62). The parkinsonian signs consist of bradykinesia, rigidity, and a prominent gait disorder; rest tremor is seen less frequently than in PD (26,64). A diagnosis of DLB may be difficult to differentiate from AD or from typical PD with dementia (PDD). However, in PDD, the interval between the onset of parkinsonism and onset of the cognitive disturbance is usually more than a year (62).
Progressive Supranuclear Palsy, Multiple System Atrophy, and Corticobasal Degeneration
PSP, MSA, and CBD closely resemble PD and may be clinically indistinguishable in the early stages of progression, but they usually emerge as distinct clinical
entities as the disease evolves. Each has a distinctive neuropathology. PSP presents as an akinetic-rigid syndrome with a prominent gait disorder early in the course of illness. In contrast, walking is usually not a major issue in PD until late in the course. Truncal posture in PSP tends to be upright instead of stooped, and axial and asymmetric limb dystonia may be present (29). Tremor is much less common in PSP than in PD (29). Cognitive impairment occurs earlier than in PD, but the dementia of PSP is usually not as severe as it is in PD, CBD, and some of the other parkinsonian syndromes. Many patients demonstrate a pseudobulbar affect, marked by uncontrollable and inappropriate laughing or crying. Several characteristic eye movement abnormalities are seen, the most important of which is a supranuclear paresis of conjugate gaze, beginning with symptomatic slowing of gaze in the vertical plane. Patients often complain of having trouble looking up, looking at their food during meals, or hitting a golf ball. Examination may reveal impaired voluntary gaze, spontaneous minimyoclonic jerks of the eyes (square wave jerks), and loss of optokinetic nystagmus, which can be elicited in the normal subject by passing a sequence of stripes or other similar repetitively occurring graphic objects horizontally or vertically across the field of vision (29). Reflex eye movements are preserved because the pathology of PSP interrupts the neural circuits descending from the centers of voluntary gaze in the frontal lobe before they damage the oculomotor nuclei in the brainstem; hence, the term “supranuclear” to the paresis of voluntary gaze in PSP. These relatively well-preserved reflexive eye movements may be demonstrated using the doll’s head maneuver, in which the patient is asked to visually fixate a target while the examiner passively moves the patient’s head in the horizontal or vertical plane. Preserved reflexive eye movements allow patients to hold their eyes on target despite the head movements. In the late stages of PSP, voluntary and reflexive eye movements in all directions, including the horizontal plane, are severely impaired. MRI may show atrophy of the dorsal midbrain (85).
entities as the disease evolves. Each has a distinctive neuropathology. PSP presents as an akinetic-rigid syndrome with a prominent gait disorder early in the course of illness. In contrast, walking is usually not a major issue in PD until late in the course. Truncal posture in PSP tends to be upright instead of stooped, and axial and asymmetric limb dystonia may be present (29). Tremor is much less common in PSP than in PD (29). Cognitive impairment occurs earlier than in PD, but the dementia of PSP is usually not as severe as it is in PD, CBD, and some of the other parkinsonian syndromes. Many patients demonstrate a pseudobulbar affect, marked by uncontrollable and inappropriate laughing or crying. Several characteristic eye movement abnormalities are seen, the most important of which is a supranuclear paresis of conjugate gaze, beginning with symptomatic slowing of gaze in the vertical plane. Patients often complain of having trouble looking up, looking at their food during meals, or hitting a golf ball. Examination may reveal impaired voluntary gaze, spontaneous minimyoclonic jerks of the eyes (square wave jerks), and loss of optokinetic nystagmus, which can be elicited in the normal subject by passing a sequence of stripes or other similar repetitively occurring graphic objects horizontally or vertically across the field of vision (29). Reflex eye movements are preserved because the pathology of PSP interrupts the neural circuits descending from the centers of voluntary gaze in the frontal lobe before they damage the oculomotor nuclei in the brainstem; hence, the term “supranuclear” to the paresis of voluntary gaze in PSP. These relatively well-preserved reflexive eye movements may be demonstrated using the doll’s head maneuver, in which the patient is asked to visually fixate a target while the examiner passively moves the patient’s head in the horizontal or vertical plane. Preserved reflexive eye movements allow patients to hold their eyes on target despite the head movements. In the late stages of PSP, voluntary and reflexive eye movements in all directions, including the horizontal plane, are severely impaired. MRI may show atrophy of the dorsal midbrain (85).
MSA is a diagnostic grouping of three disorders that are distinguished by parkinsonism, cerebellar ataxia, and autonomic insufficiency in combination with one of the three as the primary presenting deficit. Pyramidal tract deficits can also be seen as part of the mix. By convention, the most prominent neurologic theme determines the subclassification within the overarching domain of MSA, although any assortment of the four types of signs can occur in the same patient. Thus, a mix of parkinsonism and severe autonomic dysfunction (orthostatic hypotension, atonic bladder, and impotence) would be called autonomic MSA or the Shy-Drager syndrome. Pure parkinsonism is called striatonigral MSA or MSA-p; and cerebellar ataxia without a family history is called cerebellar MSA, MSA-c, or olivopontocerebellar atrophy in the old terminology (57). Cerebellar ataxia with a family history is included in the large and growing classification of the spinocerebellar ataxias (88). Autonomic failure and postural instability are early findings. As in PSP, tremor is rare. The MRI scan, which is normal in PD and in most cases of MSA, may sometimes show a combination of shrinkage of the brainstem and cerebellum and an abnormally large area of low signal in the putamen (28). The course of MSA is generally faster than that of PD. In most patients with MSA, especially the parkinsonian form, cognitive function is preserved.
CBD is characterized by highly asymmetric parkinsonism that is associated early on with cognitive impairment and pathognomonic signs that may escape the notice of the naive observer. An early and common sign of CBD is a clumsy or “useless arm” due to rigidity, dystonia, akinesia, or motor apraxia in some combination. Other early features include gait impairment, if onset is in the leg, due to lateralized stiffness or apraxia (60). In addition to dementia, other manifest higher cortical signs are ideational apraxia, aphasia, cortical sensory abnormalities (e.g., loss of spatial orientation), and alien-limb phenomenon. Additional hyperkinetic movements include coarse action tremor and myoclonus (53). Tremor is less likely to be present in CBD than in PD, but when it does manifest, it can be differentiated from Parkinson tremor because it occurs mainly with action, is coarser and higher in frequency, and is associated with marked rigidity of the affected limb or limbs. In cases of suspected CBD, MRI scan may show asymmetric shrinkage of one half of the parietofrontal cortex. The diagnosis of CBD early in its course can be difficult due to the overlap of symptoms with other neurodegenerative diseases, especially PSP, MSA, DLB, and AD.
PSP, MSA, and CBD usually respond poorly to levodopa and other anti-Parkinson drugs. However, a trial of levodopa of as much as 1,000 mg per day may be needed to exclude PD with a high response threshold. Failure to respond is strong evidence that MSA, not PD, is the probable diagnosis. The therapeutic response in patients with PSP, MSA, or CBD is not only disappointing, but the effect may be counterproductive because of aggravation of orthostatic hypotension, particularly in MSA. If patients fail to respond to levodopa, there is no reason to try other dopaminergic agents because they have less therapeutic potency and usually cause more side effects than levodopa. Botulinum toxin (BTX) injections may be useful for focal dystonia, such as eyelid opening apraxia, in PSP and MSA. Autonomic insufficiency,
particularly orthostatic hypotension, can be treated with specific drugs. Nonpharmacologic therapies, such as speech therapy, swallowing therapy for dysphagia, and physical therapy, are also an important part of patient care.
particularly orthostatic hypotension, can be treated with specific drugs. Nonpharmacologic therapies, such as speech therapy, swallowing therapy for dysphagia, and physical therapy, are also an important part of patient care.
TREATMENT OF PARKINSON DISEASE
The objective of treating PD is to suppress symptoms and improve neurologic function. No disease-modifying treatment has yet been proved to slow or reverse the underlying progression of the disease. Some therapeutic strategies, however, may avert or delay the complications that are associated with long-term dopaminergic therapy. The first decision for doctor and patient after the diagnosis is whether the symptoms require treatment. As a general rule, pharmacotherapy should be considered when symptoms begin to interfere with a patient’s ability to function at home or work. Once initiated, treatment should be individualized, taking the patient’s age, stage of disease, mental state, and tolerance of side effects into account. The algorithm shown in Figure 22-2 outlines a common approach to treatment.
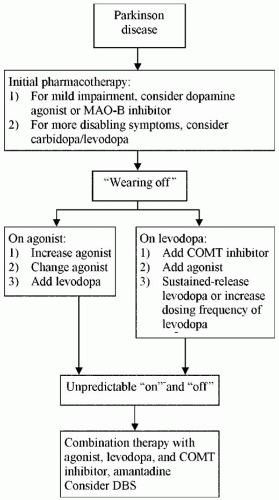 Figure 22-2. Approach to the pharmacological management of Parkinson’s disease. |
Nonpharmacologic Therapies
The care of patients with PD is best accomplished through a multidisciplinary approach, which should include access to psychological counseling; occupational, physical, and speech therapy; and support groups. Nonpharmacologic intervention can be as important as medications and should be started early in the course of PD. Aerobic and strengthening exercise is vitally important as a health maintenance routine and as a method of exerting psychological control over a chronic, variably progressive disease. Many patients benefit from the mantra, “I have PD but it doesn’t have me.”
Pharmacotherapy
Levodopa Levodopa was shown to be the first truly effective drug for treating parkinsonism 40 years ago, a distinction that has not been eclipsed by the half dozen or more drugs introduced to the marketplace since then (Table 22-2). Levodopa, the inert precursor of dopamine, was developed as an anti-Parkinson drug because it has long been known that dopamine does not cross the blood-brain barrier, whereas levodopa does. Hence, levodopa enters the brain, where it is decarboxylated by dopa decarboxylase to the therapeutically active dopamine. Levodopa works most efficiently and with fewest side effects when administered in combination with carbidopa, an inhibitor of peripheral dopa decarboxylase, which greatly diminishes the amount of “unprotected” levodopa available to cross the blood-brain barrier into the central nervous system for conversion to dopamine. Carbidopa is joined with levodopa in different ratios (10/100, 25/100, or 25/250) in short-acting and controlled-release formulations. A typical starting dose of carbidopa/levodopa (Sinemet) is one half of a 25/100-mg tablet three times a day (with meals to prevent nausea). Once tolerance is established at a low dose, the amount of carbidopa/levodopa per dose can be slowly increased, usually in half-tablet increments, to an optimal therapeutic level compatible with realistic expectations for the individual patient. Average daily consumption in early stages of disease ranges between 300 and 600 mg/day in three divided doses. The total required daily dose of levodopa tends to increase as the disease progresses, and closer spacing of doses is required to combat the “wearing-off effect” (10). Most patients with typical PD will have a gratifying and smooth initial response to levodopa. Many symptoms improve, but most patients will observe that bradykinesia improves the most. Tremor suppression often lags behind, but it, too, can be well controlled by levodopa.
One of the major drawbacks of levodopa is its short half-life of 90 minutes, which has a major impact on patient satisfaction as the disease progresses. In the
early phase of PD when patients with typical PD first begin using levodopa, the overall response is positive and smooth throughout the day. Few doses per 24 hours are required because of the brain’s ability to store the drug in the axon terminals of the still adequate supply of nigrostriatal neurons that have not been decimated by the underlying degenerative process. As more neurons degenerate, the brain’s storage capacity declines, and at some point, which varies with the individual’s inherent rate of progression, the physiologically smooth long-duration response (LDR) to levodopa gives way to the physiologically short-duration response (SDR). Clinical expression of the SDR occurs in the form of motor fluctuations, which are characterized by a “wearing off” of the benefit of a dose of carbidopa/levodopa, usually 3 to 4 hours after a dose is taken. The patient will notice a gradual re-emergence of parkinsonian symptoms during this interlude, which at its nadir is called the “off” period. The reciprocal “on” response is gradually re-established (usually
within 30 minutes) after the next dose of levodopa is swallowed and transported by gastric contraction to the absorption site in the proximal duodenum or jejunum. Levodopa-induced choreiform or dystonic involuntary movements, called “dyskinesia,” commonly begin to occur at the stage of the disease when motor fluctuations start to appear, usually at peak effect 1 to 2 hours after a dose (20). In most patients, dyskinesias are dose related and can be so severe and disabling that they greatly interfere with all activities of daily living. They can be abolished by lowering the dose of levodopa but often at the expense of the benefit provided.
early phase of PD when patients with typical PD first begin using levodopa, the overall response is positive and smooth throughout the day. Few doses per 24 hours are required because of the brain’s ability to store the drug in the axon terminals of the still adequate supply of nigrostriatal neurons that have not been decimated by the underlying degenerative process. As more neurons degenerate, the brain’s storage capacity declines, and at some point, which varies with the individual’s inherent rate of progression, the physiologically smooth long-duration response (LDR) to levodopa gives way to the physiologically short-duration response (SDR). Clinical expression of the SDR occurs in the form of motor fluctuations, which are characterized by a “wearing off” of the benefit of a dose of carbidopa/levodopa, usually 3 to 4 hours after a dose is taken. The patient will notice a gradual re-emergence of parkinsonian symptoms during this interlude, which at its nadir is called the “off” period. The reciprocal “on” response is gradually re-established (usually
within 30 minutes) after the next dose of levodopa is swallowed and transported by gastric contraction to the absorption site in the proximal duodenum or jejunum. Levodopa-induced choreiform or dystonic involuntary movements, called “dyskinesia,” commonly begin to occur at the stage of the disease when motor fluctuations start to appear, usually at peak effect 1 to 2 hours after a dose (20). In most patients, dyskinesias are dose related and can be so severe and disabling that they greatly interfere with all activities of daily living. They can be abolished by lowering the dose of levodopa but often at the expense of the benefit provided.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


