Multiple Sclerosis and other Demyelinating Diseases
Maria K. Houtchens
David M. Dawson
Background
1. Multiple sclerosis (MS) is the most common disabling neurologic condition of young adults in European and North American populations. It was first recognized as a disease entity in the latter part of the 19th century. The first description of “disseminated sclerosis” dates back to 1835 and is credited to French neurologist Cruvellhier. Jean Martin Charcot, at the Salpetriere Hospital in Paris, France, described the ataxia and oculomotor abnormalities that are often observed in younger patients. The pathologic features at autopsy, described in the first few decades of the 20th century, are now well known.
2. The cause of MS remains unknown. Theories of the cause have reflected concepts that were popular in different eras. Lesions of MS are often found close to small venules, and thrombosis of these veins was at one time thought to be important. Stress was believed by some to play a role, reflecting ideas of the psychosomatic movement of several decades ago. A search for viruses, as intact infective agents or as DNA fragments, has unsuccessfully continued for many decades. Epstein-Barr Virus (EBV) may play a particular role in MS: The studies have found that antibodies to the viral proteins, Epstein-Barr nuclear antigens, viral capsid antigens, and diffuse early antigens are significantly raised in people with MS. One study found that people with the highest levels of antibodies to EBV were 33 times more likely to develop MS than people with the lowest levels. It is not clear whether there is a causative or temporal association between infection with a specific pathogen and MS onset. In the last few decades, genetic predisposition was determined to clearly pay a role. We now define MS as a T cell-mediated autoimmune central nervous system (CNS) disease triggered by unknown exogenous agents, such as viruses or bacteria, in subjects with a specific genetic background.
3. In most patients, MS is a chronic disease. In 85% of patients, it begins with a focal inflammatory lesion of the nervous system, developing over days and recovering after months. Further lesions develop and cause clinical relapses, usually at a rate of one or two relapses per year. Magnetic resonance imaging (MRI) data have shown us that in actuality lesions occur in the brain and spinal cord at a far more rapid pace, often 10 times as frequently as relapses that are clinically recognized. After a number of years, or even decades, most patients enter a slowly progressive phase of the illness, with increasing disability. Impairment of gait, reduced visual acuity, paresthesias and pain, loss of bladder control, and cognitive deficits dominate the clinical picture after the progressive phase has advanced further. In large registries of patients, for example, from France and from Denmark, it is found that reduction in life span due to MS is not common, but that most (75% to 80%) of the patients are disabled and unable to work by age 65.
4. Other variants of MS occur. About 10% of patients have primary progressive MS (PPMS) (i.e., no relapses are recognized and the patient steadily worsens from
the onset). Another 10% have so-called benign MS, with few relapses and no disability even though they have been known to have the disease for many decades. A small number of patients have acute MS, with frequent and large lesions and poor recovery, and it is among this group that a fatal outcome is occasionally seen.
5. Most of the data regarding treatment of MS are derived from studies that exclude variant forms of MS and instead use the more common version of the disease, with relapses followed in time by a secondary progressive phase. Since the variant forms have not been tested, it is often difficult clinically to decide whether a particular form of treatment is appropriate for an individual patient. Wide variations in the course of the disease make it imperative that carefully designed clinical trials furnish the evidence for treatment.
Epidemiology
1. MS is the most common neurologic disease among young adults.
2. Incidence is the highest from ages 20 to 40, but the disease can start even in childhood or after age 60.
3. In the United States alone, there are about 500,000 MS patients and about 10,000 new cases are diagnosed yearly.
4. There is 7:3 female-to-male ratio.
5. There are zones of high incidence and medium incidence, and there are places in the world where the disease is almost unknown.
a. Prevalence decreases with proximity to equator creating a so-called “North-South Gradient” of MS distribution.
b. High incidence includes all of Europe, North America, New Zealand, and southern Australia. In these areas, the prevalence is about 60/100,000. In Minnesota and many of the northeastern states of the United States, one person of every 500 has MS (i.e., a prevalence of 200/100,000). In general, MS is more than twice as common in the northern tier of the United States as in the southern states.
c. Race plays an important role: U.S. residents who are of Japanese, Native American, or Sub-Saharan African descent have a much lower incidence of MS than do people of Irish, British, or Scandinavian background under equal geographic circumstances.
d. Incidence of MS in African Americans is 25% of that of persons with Caucasian background. However, the disease tends to be more rapidly disabling and resistant to therapies in this patient population.
6. If persons with ethnically and geographically low risk develop MS, the disease may be atypical in clinical manifestations and imaging findings.
Pathophysiology
1. The pathologic hallmarks of MS are demyelination and predominantly perivenular inflammation. Severe or advanced disease causes axonal disruption and loss and cortical atrophy leading to a process of neurodegeneration.
2. Historically, MS was considered a disease of cerebral white matter. Recent data provide evidence for primary involvement and neurodegeneration of central and cortical gray matter.
3. The immunologic mechanism involves activation of autoreactive CD4+ cells in the peripheral immune system followed by their migration into the CNS via a disrupted blood-brain barrier (BBB). This is followed by reactivation of the cells by in situ myelin antigens, activation of B cells and macrophages, and secretion of proinflammatory cytokines and antibodies.
4. The typical lesion of MS is a few millimeters to a centimeter in size. Viewed threedimensionally, a lesion is often ovoid or linear rather than circular. This is a
feature of the MRI appearance. Activated T cells and macrophages are present. The cells express T helper 1 (TH1) cytokines such as interferon gamma (IFN-γ), tumor necrosis factor (TNF), and interleukin-2 (IL-2). Cytokines of 2the TH2 series such as IL-4, IL-10, and IL-13 are reduced. Many kinds of proinflammatory molecules, such as integrins and other adhesion molecules, are upregulated.
a. Microscopically, lesions show destruction, swelling or fragmentation of myelin sheaths, proliferation of glial cells, variable axonal destruction (new and old plaques), and variable damage to neurons, but relatively good preservation of background structure, and cystic lesions are rare.
b. Early/acute lesions (days to weeks) show marked hypercellularity, macrophage infiltration, astrocytosis, perivenous inflammation with plasma cells and lymphocytes, and disintegration of myelin.
c. Active/non-acute lesions (weeks to months) show lipid-laden phagocytes with minimal inflammatory response at the center of lesions but prominent at the edges of lesions with increased numbers of macrophages, lymphocytes, and plasma cells.
d. Chronic inactive plaques (months to years) show prominent demyelination (almost complete loss of oligodendrocytes), extensive gliosis, and hypocellularity.
e. Remyelinating plaques may result from differentiation of precursor cells common to type II astrocytes and oligodendrocytes. They show uniform areas of aberrant and incomplete myelination (shadow plaques).
5. Chronic lesions with poor recovery have the appearance on biopsy, at autopsy, or on MRI of an empty astroglial scar. The term multiple sclerosis refers to these late-stage discolored plaques or scars.
6. In demyelinated areas, transmission of nerve impulses is blocked and signals fail to arrive at their destination.
Genetics
1. If a mother has MS, her children also have a 3% to 5% chance of having MS—at least a 20-fold increase.
2. If a father has MS, his son has a 1% chance, and his daughter a 2% chance, of having MS.
3. A sibling of an affected person, including a nonidentical twin, has a 3% to 4% chance of having MS.
4. An identical twin has a 30% chance of having MS if one includes asymptomatic twins with only MRI or spinal fluid findings.
5. MS is associated with major histocompatibility complex II (MHC-II) and three specific alleles in the DR2 haplotype.
6. Full-scale genome screens have shown no convincing locus for an “MS gene.” It is likely that a number of genes contribute liability by increasing immune reactivity to common viruses or to antigenic components of myelin to which other persons are nonreactive.
Diagnosis
1. Clinically isolated syndrome (CIS), that is, optic neuritis (ON), transverse myelitis (TM), or a brainstem syndrome as the first ever episode of neurologic dysfunction. The patient does not meet criteria for MS diagnosis. Specific clinical syndrome depends on location of lesion(s) within brain, spinal cord, or optic nerves. The attack typically progresses for several days, plateaus, and then improves over days, weeks, or rarely months. Improvement can be complete, or partial.
2. Most common presenting symptoms of MS include visual/oculomotor problems (49%), leg paresis/paresthesias (42%), cerebellar ataxia (24%). Other symptoms may include progressive or abrupt cognitive changes, Lhermitte’s phenomenon (electrical painful paresthesias induced by neck flexion), Uthoff phenomenon (worsening symptoms with increased body temperature), neuropathic pain, and fatigue.
3. Clinically definite MS (CDMS): Patient meets McDonald criteria for MS diagnosis.
4. McDonald MS criteria for MS diagnosis (2001-2006) One attack with objective evidence of neurologic disease, plus a second attack, which can be defined by MRI criteria, positive spinal fluid findings, or abnormal evoked potentials. Eighty-three percent sensitive at 1 year and 83% specific at 3 years for diagnosis of MS.
McDonald MRI criteria:
Must have at least three of the following to count as dissemination in space:
• One Gd-enhancing lesion or nine T2/FLAIR hyperintensities
• One or more infratentorial lesions
• One or more juxtacortical lesions
• Three or more periventricular lesions
Dissemination in time
• Three-month follow-up MRI shows enhancement at a new site.
OR
• Six-month follow-up MRI shows new enhancing lesion or a new T-2 lesion.
5. Over the course of the disease, each attack may leave some residual deficits. Accumulation of such deficits results in increasing disability. After several attacks of various types, a patient may present with common “fixed” problems:
a. Mild reduction in vision in one eye
b. Dysconjugate eye movements, with diplopia
c. Extensor plantar responses and inability to walk heel-and-toe
d. Reduced vibration sense in the legs
e. Urgency of bladder function
f. Cognitive impairment
6. Common late-stage deficits include dementia, inability to stand or walk, slurred speech, ataxia, incontinence, and marked sensory loss in hands and legs.
Diagnostic Testing
Magnetic Resonance Imaging
1. MRI is now the dominant laboratory method for diagnosis of MS. MS lesions are usually easily detected and often are characteristic. Conventional MRI techniques are now widely accessible to community and academic neurologists. By scan:
a. Lesions are bright on T2-weighted and fluid-attenuation inversion recovery (FLAIR) images, indicating a higher than normal water content. These MRI sequences reflect the total burden of disease (Fig. 7-1A).
b. Lesions are usually isodense on T1-weighted images, indicating that the tissue itself is intact.
c. Lesions may be hypodense on T1-weighted images, indicating underlying axonal disruption (black holes) (Fig. 7-1B).
d. Lesions may be present in many areas of the brain, but most typically they are found adjacent to the lateral ventricles, oriented perpendicular to them, and in the corpus callosum (best seen on midline sagittal FLAIR images [Fig. 7-2]), and in the cerebellar peduncles. MS plaques directly touch the ventricular wall following the location of the small venules. In contrast, small vascular lesions are usually seen several millimeters away from the ventricular wall.
e. Acute and subacute lesions (<8 weeks since formation) often show enhancement on T1 postgadolinium contrast sequences indicating inflammation, BBB disruption, and recent disease activity (Fig. 7-3).
f. Size of cerebral lesions varies from 5-10 to 100 mm or greater.
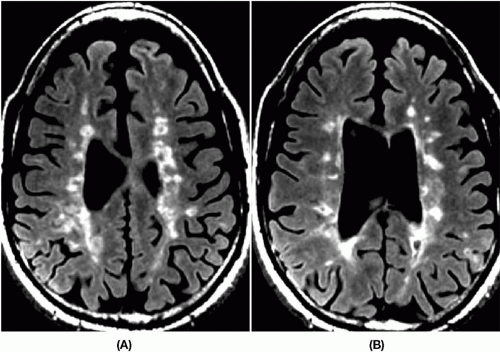
Figure 7-1. A: Axial FLAIR. This MRI was performed in a patient with secondary progressive MS. There are innumerable ovoid hyperintense lesions abutting lateral ventricles, involving the U-fibers and subcortical white matter. Cerebral atrophy is apparent: lateral ventricles are enlarged, and cortical ribbon is thinned. B: “Black holes.” T1 sequence shows hypointense lesions corresponding to some hyperintense lesions on FLAIR sequence. These are areas where severe axonal damage is seen pathologically. There is a correlation between axonal damage and neurologic disability.
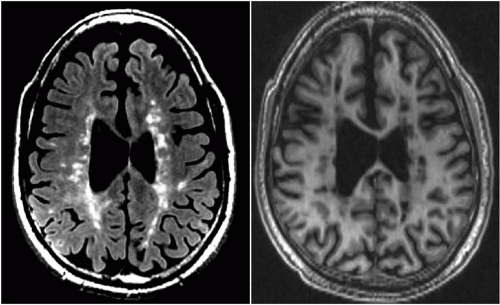
Figure 7-2. Sagittal FLAIR and T1. Typical “Dawson fingers” corresponding to areas of demyelination and inflammation are seen on sagittal FLAIR image. Atrophy of the corpus callosum is best appreciated on sagittal T1 precontrast sequence.
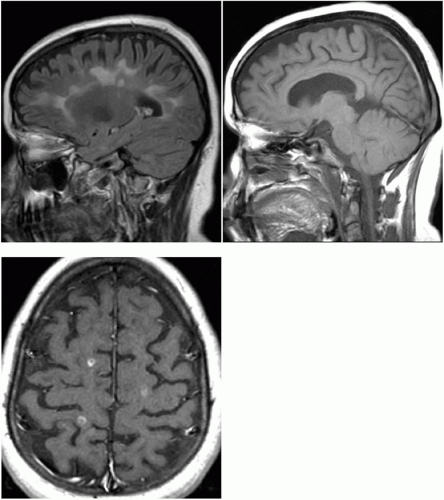
Figure 7-3. T1 postcontrast sequence. “Active” MS lesions are seen in this T1 postcontrast sequence. They appear as partially ring-enhancing or diffusely enhancing lesions. Pathologically, they correspond to areas of impaired blood-brain barrier.
g. Lesions are common in the spinal cord, especially the cervical cord opposite the C-2 or C-3 vertebrae. They typically involve less than two contiguous segments of the spinal cord and less than half of the transverse diameter of the cord.
2. Most patients with MS have MRI scan findings that are characteristic of MS. Some may have atypical or nonspecific patterns of lesions. Only extremely rarely do patients with a typical clinical course suggestive of MS have normal MRI scans. Such patients present great diagnostic difficulty, and repeated scanning and other examinations may be required.
3. Nonconventional MRI techniques are employed in clinical trials and can be used in specialized MS centers for research or disease monitoring purposes.
a. Global and focal cerebral atrophy measures in brain and spinal cord: Atrophy correlates with axonal and neuronal loss, and physical and cognitive impairment.
b. NAA (N-acetylaspartate) levels measured with MR spectroscopy: A marker of neuronal and axonal metabolism, NAA is decreased in MS lesions and in normal-appearing white matter in brains of MS patients.
c. Magnetization transfer imaging or ratio: Abnormal in more severe lesions with greater tissue destruction, it can be abnormal despite normal routine MRI sequences.
d. Functional MRI (fMRI) measures critical circuitry involved in response to injury, activation, loss of function, and recovery of function in MS.
Other Tests
1. Lumbar puncture is needed in some patients with MS, but is not performed routinely in cases of diagnostic certainty. Characteristic findings in the cerebrospinal fluid (CSF) in MS are a modest number of lymphocytes (fewer than 50/mm3), total protein less than 0.8 g/L, elevated immunoglobulin G (IgG) synthesis levels (3.3 mg/day in 90% of patients), and high IgG index (0.7 or greater in 90% of patients). Myelin basic protein (MBP) is normally <1 ng/mL but increases in 80% of acute MS relapses. Presence of oligoclonal banding (OCB) on electrophoresis is the most sensitive of the CSF tests, being present in 75% to 80% of patients with established MS, and in 50% to 60% of patients with CIS. OCB may also be present in other infectious/autoimmune conditions such as Lyme disease, neurosarcoidosis, neurosyphilis, and human immunodeficiency virus (HIV).
2. Evoked potentials testing—especially testing of visual evoked potentials—will occasionally help. It can establish evidence of prior damage to optic nerves in the absence of a clear clinical history by showing unilateral prolongation of P100 wave.
Differential Diagnosis
Many other neurologic conditions may be confused with MS (Table 7-1). They fall into two categories:
1. Diseases that look like MS clinically, including other CNS inflammatory diseases such as lupus, sarcoidosis, and chronic meningitis, and degenerative processes such as hereditary ataxia, adrenoleukodystrophy, and motor neuron disease
2. Diseases that look like MS by MRI findings, including other causes of “white spots”
a. Vascular disease: Small-vessel disease in hypertension, migraine, CADASIL (cerebral autosomal dominant anteriopathy with subcortical infarcts and leukoencephalopathy).
b. Infections: Lyme disease, HIV.
TABLE 7-1 Diagnoses That Mimic Multiple Sclerosis
Infectious
Toxic-metabolic
Inflammatory
Other
Lyme disease
B12 deficiency
SLE
CADASIL
Neurosyphilis
Vitamin E deficiency
Sjögren disease
CNS lymphoma
HIV
Neuro-Behçet disease
Cerebrovascular disease
HTLV-1
Sarcoidosis
Leukodystrophies
PML
CNS vasculitis
Motor neuron disease
Cervical spondylosis
HIV, human immunodeficiency virus; HTLV, human T-lymphocyte virus; PML, leukoencephalopathy; SLE, systemic lupus erythematosus; CADASIL, cerebral autosomal dominant anteriopathy with subcortical infarcts and leukoencephalopathy; CNS, central nervous system.
c. Granulomatous disease: Sarcoidosis, Behçet disease
d. Monosymptomatic demyelinating disease: TM and acute disseminated encephalomyelitis (ADEM)
Classification and Clinical Considerations
1. Several MS classifications are used.
a. Based on disability accumulation: Benign MS (5% of all patients)—no or minimal neurologic disability after 10 to 15 years. Malignant MS (5% to 7% of all patients)—neurologic disability requiring ambulation assistance after ≤5 years.
b. Based on clinical course: Relapsing-remitting MS (RRMS). This subtype is the most common (85% of all patients fit into this disease category at diagnosis). It is characterized by relapses and remissions of neurologic disability over years to decades. Incomplete recovery from relapses often leads to disability accumulation. Secondary progressive MS (SPMS) follows RRMS in 10 to 25 years after the diagnosis in 60% to 80% of patients. This subtype is characterized by absence of relapses and progressive worsening of neurologic function involving the pyramidal system, cerebellar connections, dorsal columns, and cortical association fibers. Patients exhibit paraparesis or hemiparesis, ataxia of gait, sensory ataxia, neuropathic pain symptoms, and cognitive decline. Ambulation assistance is often required at this stage. PPMS often presents with indolent or rapid evolution of neurologic symptoms and usually involves progressive leg weakness with difficulty walking as the initial feature of the disease. Careful history taking confirms absence of exacerbations of neurologic deficits. This disease category is more common in men, in the fourth and fifth decades of life. Prognosis is worse for this group of patients. They do not respond to currently available MS therapies.
c. Based on predominant clinical subtype: In recent years, specific clinical phenotypes of MS are identified based on the most affected neurologic subsystem. These include “spinal MS variant,” “cerebellar MS variant,” and “cognitive MS variant.”
2. The combination of several epidemiologic, clinical, and imaging factors carry better prognosis for stable disease course. Positive prognostic factors include
a. Younger age of onset
b. Female sex
c. Monosymptomatic onset
d. Sensory symptoms or ON at onset
e. Few T2 of FLAIR lesions on original MRI




