and Aditya G. Shivane1
(1)
Cellular and Anatomical Pathology Level 4, Derriford Hospital, Plymouth, UK
Abstract
Muscle diseases are relatively uncommon disorders and often require muscle biopsy to aid diagnosis. This chapter describes how a muscle biopsy should be taken and dealt with within the laboratory, outlining the special stains required to classify the type of muscle disease. The pathology of different types of muscle disease is described along with a detailed classification of disorders of muscle and, in genetic forms of muscle disease, the underlying genetic defects are described.
Keywords
Muscle biopsyDystrophyHistochemistryMyositisMyopathyPrimary muscle diseases are relatively uncommon and generally dealt with by neurologists or physicians with an interest in muscle disease. As part of the diagnostic work up, a muscle biopsy may be helpful in trying to confirm the nature of the muscle disease. However, it is important that the biopsy procedure is done correctly to maximise the information obtained from the biopsy, and that the specimen is examined in a specialist neuropathology or muscle pathology laboratory.
In order for the appropriate tests to be undertaken on the biopsy, and for the pathologist to interpret the findings correctly, it is essential that the clinician looking after the patient provides adequate information to the pathologist. This should include presenting symptoms and examination findings (including distribution of weakness, the presence of wasting or hypertrophy), medication history (including duration and dose of any immunosuppressive treatment), the presence or absence of a family history of neuromuscular disease, cardiac, ocular or nervous system involvement, EMG findings and creatine kinase levels. In addition, in children the age at which developmental milestones were achieved and functional level are important. MRI examination of muscle can be useful in demonstrating patterns of muscle involvement and identifying a target for biopsy.
9.1 Muscle Biopsy Technique
Prior to taking the muscle it is important that the clinician liaises with the surgeon to ensure that the correct muscle is biopsied. The laboratory should be informed of the timing of the biopsy so that they can prepare the relevant reagents that are needed to snap freeze the sample on arrival. The muscle should be clinically affected, but not so severely that the biopsy would only reveal end stage disease. The sample should be taken from the belly of the muscle, avoiding fascial and tendon insertions. Most muscle diseases affect proximal muscles and commonly sampled muscles include deltoid, quadriceps femoris, biceps brachialis, tibialis anterior and gastrocnemius. Virtually all muscle biopsies may be taken under local anaesthesia (although care must be taken not to infiltrate local anaesthetic within the muscle fascia), but younger children and some anxious adults may require sedation. General anaesthesia carries significant additional risk, and in a number of muscle disorders, has the particular risk of malignant hyperthermia.
Muscle biopsies may be undertaken using either a conchotome, or more commonly a needle (either Edwards or Bergstrom), 4–6 mm in diameter. These techniques result in a smaller sample than with an open biopsy, however, patients can normally resume normal activities almost immediately after the procedure. An open biopsy will result in a scar approximately 3 cm in length, but allows a number of samples to be taken through the same incision and generally a better sized sample. We prefer open biopsies and generally take 3 samples measuring 10 × 5 × 5 mm each, along the long axis of the muscle. Open biopsies can be orientated by laboratory staff more easily than needle or conchotome samples, facilitating histological assessment. Clamping of the muscle between two sets of forceps prior to removal can be undertaken with an open procedure to reduce contraction artefact in samples used for electron microscopy. However, we have not found that clamping is necessary, and contraction artefact can be minimised by careful laboratory handling of the sample.
9.2 Laboratory Preparation
Muscle biopsies should be handled in a specialist neuropathology/muscle pathology laboratory. Ideally the sample should be received in the laboratory within 20 min of removal, but if necessary (e.g. transfer between hospitals) samples can be transported in a chamber kept moist with normal saline, and chilled by ice (not in direct contact with the muscle sample), for up to 2 hr. Although glycogen is rapidly depleted, most of the other stains and histochemical preparations are retained reasonably well. Once in the laboratory, the sample needs to be orientated correctly in order to allow a transverse section across the long axes of the fibres and it is frozen in isopentane which has been cooled in liquid nitrogen to −160 °C. Once frozen, sections can be cut within a cryostat for routine staining (Table 9.1), histochemical preparations demonstrating muscle enzyme activity (Table 9.2) and immunohistochemistry to detect specific proteins in tissue (Table 9.3). A small amount of tissue should be fixed in glutaraldehyde for electron microscopy and some tissue kept frozen to allow further analysis of the sample, for example Western blot, DNA analysis or enzyme assays. Formalin fixed paraffin sections are of limited value, but may be helpful in the diagnosis of vasculitis.
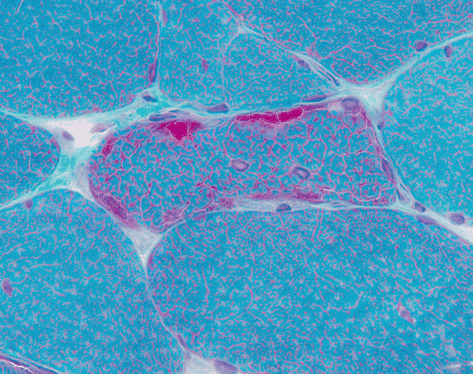
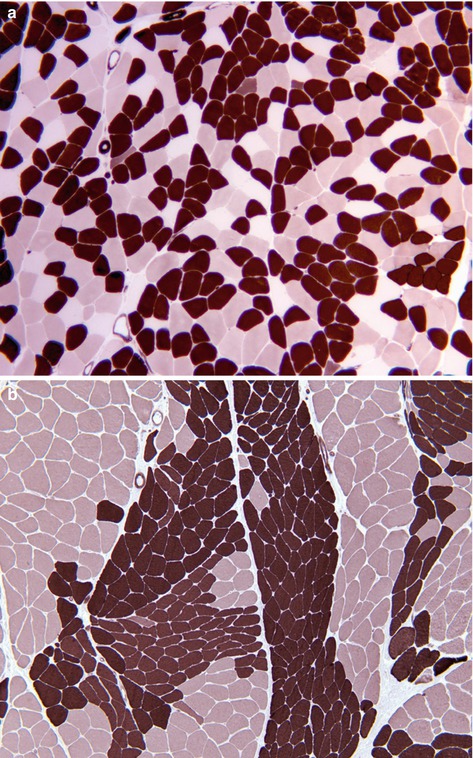
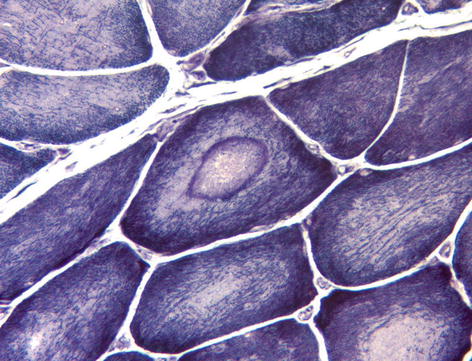
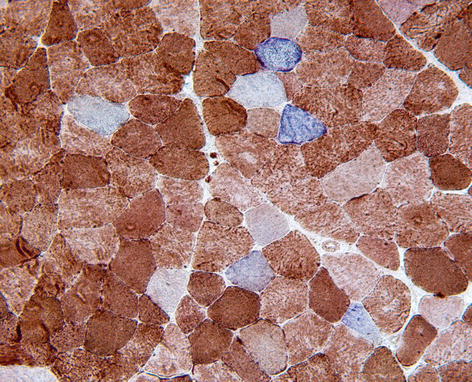
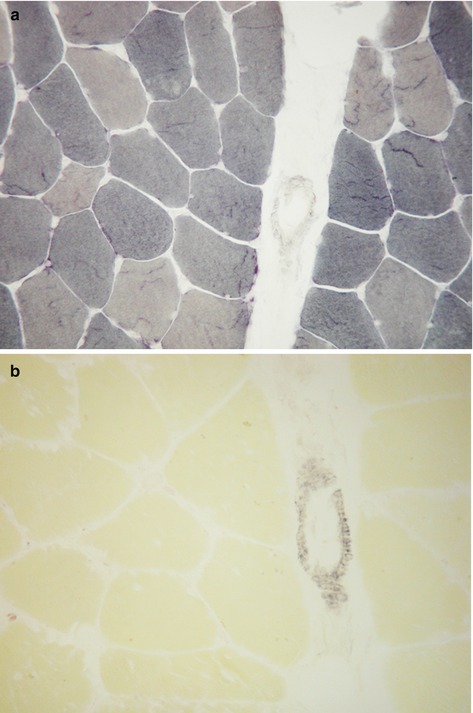
Table 9.1
Histological stains
Haematoxylin and eosin (H&E) | General pathological reactions such as fibre necrosis, regeneration, hypertrophy, atrophy, inflammation and vasculitis. |
Periodic acid Schiff (PAS) | Demonstrates glycogen, which accumulates in glycogen storage diseases, steroid myopathy, hypothyroid myopathy. |
Oil-red-O or Sudan black | Demonstrates neutral lipids, which accumulate in lipid disorders, alcoholic myopathy and steroid myopathy. |
van Gieson (VG) | Demonstrates collagen which increases in many chronic muscle disorders, particularly the muscular dystrophies. |
Gomori-trichrome | Muscle fibres appear green, mitochondria and some cytoplasmic bodies (e.g. nemaline rods) appear red. ‘Ragged-red’ fibres are seen in mitochondrial myopathies (Fig. 9.1). |
Congo-red | Demonstrates amyloid. Modified version is helpful in identifying inclusions of inclusion body myositis. |
Table 9.2
Histochemical preparations
Myosin adenosine triphosphatase (ATPase) | Demonstrates different fibre types by varying the pH of reaction. At acid pH type 1 fibres are reactive, and in alkaline pH type 2 fibres are reactive. Type 2 fibres can be further distinguished into 2a and 2b in acid pH (Fig. 9.2a). In some muscle disorders only one fibre type may be affected, and groups of each fibre type indicates reinnervation following denervation (Fig. 9.2b). |
Reduced nicotinamide adenine dinucleotide tetrazolium reductase (NADH-TR) | An oxidative enzyme present on the sarcoplasmic reticulum and within mitochondria. Useful as a sensitive marker of internal fibre architecture which is disrupted in many myopathies. Also highlights tubular aggregates and, in denervation, the atrophic fibres stain darkly and may develop a targetoid appearance (Fig. 9.3). |
Succinate dehydrogenase (SDH) | An oxidative enzyme specific to mitochondria. Useful as an indicator of mitochondrial numbers which may accumulate (or rarely be depleted) in mitochondrial disorders. |
Cytochrome oxidase (COX) | Oxidative enzyme present within mitochondria, partly encoded by mitochondrial DNA. Segments of muscle fibres are deficient of COX in mitochondrial disorders, and to a lesser extent in aging. Preparation may be combined with SDH (Fig. 9.4). |
Myophosphorylase | Glycolytic enzyme which is absent within muscle fibres in McArdle’s disease (Fig. 9.5). |
Myoadenylate deaminase | Enzyme that is deficient in myoadenylate deaminase deficiency. |
Phosphofructokinase | Glycolytic enzyme absent in type VII glycogenosis. |
Acid phosphatase | Lysosomal enzyme that accumulates in lysosomal storage disorders, vacuolar myopathies and acid maltase deficiency. |
Table 9.3
Antibodies used for immunohistochemistry
Human leucocyte antigen class 1 (HLA-1) | Increased sarcolemmal and sarcoplasmic expression in inflammatory disorders of the muscle, including in areas of the biopsy distant from inflammatory cells. |
p62 (sequestosome-1) | Useful in identifying inclusions within muscle fibres, including those seen in myofibrillar myopathies and inclusion body myositis. |
Membrane attack complex (complement C5b-9) | Increased capillary expression in dermatomyositis and anti-synthetase myositis. Perimysial and fascial expression may also be seen. |
CD3 and CD20 | Useful in identifying T and B lymphocytes respectively in the assessment of inflammatory muscle disease |
Dystrophin | Deficient in dystrophin-related muscular dystrophies |
Sarcoglycans | Deficient in LGMD 2C-2F |
Dysferlin | Deficient in LGMD 2B/Miyoshi myopathy |
Caveolin-3 | Deficient in LGMD 1a, rippling muscle disease and hyperCKemia |
Laminin alpha 2 or merosin | Deficient in merosin deficient congenital muscular dystrophy |
Collagen type VI | Deficient in Ullrich congenital muscular dystrophy |
Emerin | Deficient in X-linked Emery Dreifuss muscular dystrophy |
SERCA1 | Deficient in Brody’s disease |
Myotilin and desmin | Accumulate in myofibrillar myopathies |
Telethonin | Deficient in LGMD 2G |
Plectin | Deficient in LGMD 2Q |
Lysosome-associated membrane protein, LAMP-2 | Deficient in Danon’s disease |
Actin | Accumulates in nemaline rods |
Fig. 9.1
Muscle showing ‘ragged-red’ fibre with accumulation of red staining mitochondria. Gomori-trichrome preparation. These are characteristic of mitochondrial disorders, but may occasionally be seen in normal aging and chronic myopathies such as IBM
Fig. 9.2
(a) Random ‘checker board’ pattern of type 1, 2a and 2b fibres in normal muscle. (b) Muscle showing groups of both type 1 and type 2 fibres, which is pathognomonic of reinnervation following denervation. ATPase histochemistry
Fig. 9.3
Muscle fibre showing central ‘bulls eye’ appearance (target fibre) characteristic of denervation. NADH histochemistry
Fig. 9.4
Cytochrome oxidase-negative/SDH-positive fibres, which appear blue in muscle biopsy. These are a sensitive marker of mitochondrial disorders, but may also be seen in smaller numbers in normal aging and chronic myopathies, such as IBM. COX/SDH histochemistry
Fig. 9.5
(a) Myophosphorylase histochemistry showing normal enzyme activity in muscle fibres. (b) Absent enzyme activity in skeletal muscle fibres (but not smooth muscle of blood vessels) in McArdle’s disease
There are a number of additional glycolytic enzymes (e.g. Phosphorylase B-kinase, phosphoglycerate kinase, phosphoglycerate mutase and lactate dehydrogenase) which may be assessed biochemically in muscle.
The sample fixed in glutaraldehyde for electron microscopy, is post fixed in osmium tetroxide, and then processed into resin prior to being stained with uranyl acetate and lead citrate. This allows fine ultrastructural examination of the muscle fibre internal architecture and visualisation of myofibrils, mitochondria and other internal structures. Ultrastructural examination is important in the diagnosis of a number of muscle disorders where there are characteristic structural abnormalities e.g. congenital myopathies, mitochondrial myopathies, inclusion body myositis.
9.3 Normal Structure of Muscle
Some basic muscle histology is covered in Chap. 1. Muscle fibres are syncytia formed by the fusion of foetal myoblasts, and are arranged into groups or fascicles surrounded by a fine layer of collagen (the perimysium). Muscle fibres may extend for several centimetres in length and in transverse section have a hexagonal outline, usually with 4–6 subsarcolemmal nuclei visible in each histological section (see Chap. 1). Muscle fibre diameters (minimum Ferets) range from 40 to 80 μm in adult males and 30 to 70 μm in adult females, but are smaller in infants and children. In adults, muscle fibres can be divided into three major groups; type 1 fibres – slow twitch oxidative, type 2a – fast twitch oxidative and type 2b fibres – fast twitch glycolytic. These fibre types can be distinguished most easily using the ATPase preparations at different pHs (see above) or by immunohistochemistry using antibodies to slow and fast myosin or SERCA 1 and 2. Glycolytic fibres contain more glycogen and glycolytic enzymes, whereas oxidative fibres rely more on lipid, oxidative enzymes and contain relatively more mitochondria. The fibres are arranged into groups innervated by a motor neuron (motor unit), which may range in size from a handful of fibres in an extraocular muscle, to several hundred fibres in a limb muscle. All fibres from each individual motor neuron are of the same fibre type, however, they are randomly intermingled during muscle development producing a ‘checkerboard’ pattern on ATPase preparations (see Chap. 1). The proportions of different fibre types varies considerably between different muscles depending on the function of the muscle, and also between the deep and superficial aspects of the muscle. Therefore, interpretation of changes in muscle fibre type distribution requires a knowledge of the normal range for that particular muscle. In the superficial parts quadriceps femoris, there are approximately equal numbers of type 1, type 2a and type 2b fibres. Muscle spindles are sensory receptors that consist of specialised intrafusal muscle fibres within a fibrous capsule and measure fibre tension and length.
9.4 General Pathological Reactions
Muscle may show neurogenic or myopathic types of pathological change, and in some cases both may be present. Following denervation the most striking finding is that of fibre atrophy (Fig. 9.6), often with flattened, angular, atrophic fibres eventually resulting in small clusters of nuclei with little visible cytoplasm (pyknotic nuclear clumps). Occasional necrotic and regenerating fibres may also be seen in muscles showing neurogenic atrophy, and a helpful finding is the presence of darkly stained atrophic fibres with an NADH-TR preparation and, less commonly, fibres having a target appearance (Fig. 9.3). In cases where there is reinnervation, areas composed of groups of each fibre type (fibre type grouping) may be seen along with grouped atrophy (Fig. 9.2). In slowly progressive chronic denervating conditions secondary myopathic change may occur, which in some cases may resemble a muscular dystrophy.
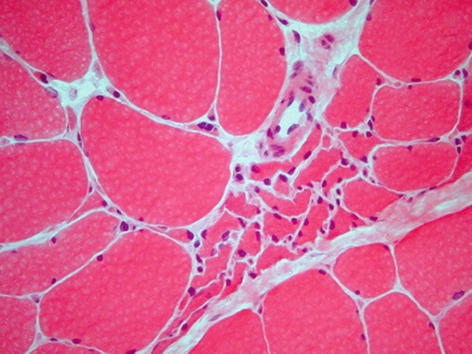
Fig. 9.6
Chronic denervation showing scattered angular atrophic fibres and central group of atrophic fibres. H&E stain
General features of a myopathic disorder include an increase in variation of fibre size, increased numbers of internal nuclei, fibre necrosis, regeneration and fibroadipose replacement of muscle (Fig. 9.7). In certain chronic muscular disorders, particularly muscular dystrophies, fibre hypertrophy associated with splitting is seen. Type 1 fibre predominance and atrophy may be seen in congenital myopathies. Other general chronic myopathic changes include disordered fibre architecture including whorled, lobulated and ring fibres. Inflammatory cell infiltrates are a feature of myositis, but may also be prominent in some cases of muscular dystrophy.
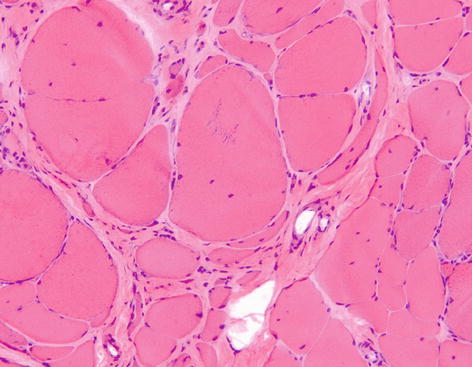
Fig. 9.7
Muscle biopsy showing chronic myopathic change with marked fibre hypertrophy and atrophy, increased numbers of fibres with central nuclei and increased connective tissue between fibres H&E stain
Most muscle diseases can be divided into the following groups: muscular dystrophies, congenital myopathies, myofibrillar myopathies, metabolic myopathies, channelopathies, inflammatory myopathies and toxic/drug induced myopathies. However, this classification is being challenged by the recent expansion of genetic changes associated with muscle disease, as there is significant overlap between these groups, with several gene defects resulting in different clinical disorders and vice versa.
9.5 Muscular Dystrophies
The muscular dystrophies are inherited disorders which predominantly affect skeletal muscle, that are usually progressive and diagnosed in childhood or early adult life. Many affect proximal muscle groups, but some have a more restrictive pattern of muscle involvement. Some muscular dystrophies also affect cardiac muscle and the central nervous system. In most patients the serum creatine kinase is markedly elevated.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


