Chapter 10 Muscle disorders
Introduction
Muscle diseases include a number of rare, often progressive conditions leading to physical disability and, frequently, reduced life expectancy. Although each condition is rare, the overall prevalence of muscle disease is 1:1000 and individual disorders tend to present at around the same age, although there may be a wide range. Significant improvements in management means that affected children are surviving to middle age where previously they would have died and they are now presenting for care in adulthood (Wagner et al., 2007). Knowledge of these conditions tends to be poor outside the specialist area of paediatric physiotherapy. This chapter gives an overview of the most frequently seen disorders so the physiotherapist is aware of the different diagnoses which can influence overall management. For many conditions, management strategies are transferable and knowledge of the most frequently seen conditions will be relevant even to the most rare disorder. For a comprehensive text on muscle disorders the reader is referred to Dubowitz (1995).
Classification and diagnostic investigations
Since the 1980s tremendous advances in genetics and molecular biology have enhanced our understanding of the pathogenesis of muscle disorders. This has led to the availability of genetic testing for many conditions which has broadened our concept of classical phenotypes to include milder, even subclinical presentations. Becker muscular dystrophy (BMD) is a good example, whereby the disorder can result in loss of mobility as early as 16 years of age or as late as the sixth decade (Bradley et al., 1978), yet some patients present only with exertional cramps and myoglobinuria (Gospe et al., 1989). The limb girdle muscular dystrophies (LGMD) are another example. They are a heterogeneous group of disorders classified according to age of onset, inheritance pattern, and molecular and genetic defect, as shown in Table 10.1. The classification of congenital muscular dystrophies (CMD), based on clinical and/or pathological features, is also expanding and several forms are alleic with LGMD as shown in Table 10.2.
Table 10.1 Classification of the limb girdle and sex-linked muscular dystrophies in relation to the genetic defect
| Type | Gene Locus | Gene Product |
|---|---|---|
| Autosomal dominant | ||
| LGMD1A | 5q22-q34 | Myotilin |
| LGMD1B | 1q11-21 | Lamin A/C |
| ADEDMD | 1q11-21 | Lamin A/C |
| LGMD1C | 3p25 | Caveolin 3 |
| Autosomal recessive | ||
| LGMD2A | 15q15.1-q21.1 | Calpain 3 |
| LGMD2B | 2p13 | Dysferlin |
| LGMD2C | 13q12 | γ Sarcoglycan |
| LGMD2D | 17q21 | α Sarcoglycan |
| LGMD2E | 4q12 | β Sarcoglycan |
| LGMD2F | 5q33-q34 | δ Sarcoglycan |
| LGMD2G | 17q11-12 | Telethonin |
| LGMD2H | 9q31-34.1 | Not yet known |
| LGMD2I | 19q13.3 | FKRP |
| LGMD2J | 2q31 | Titin |
| LGMD2K | 9q34.1 | POMT1 |
| LGMD2L | 11p13-p12 | |
| LGMD2M | 9q31 | POMT2 |
| Sex-linked | ||
| Dystrophinopathy | Xp21 | Dystrophin |
| (DMD, BMD) | ||
| EDMD | Xq28 | Emerin |
LGMD, limb girdle muscular dystrophy; ADEDMD, autosomal dominant Emery Dreifuss muscular dystrophy; DMD, Duchenne muscular dystrophy; BMD, Becker muscular dystrophy; EDMD, Emery Dreifuss muscular dystrophy.
The severity of respiratory muscle involvement does not always correlate with the severity of skeletal weakness and it is, therefore, essential that all patients with neuromuscular disorders are monitored regularly for forced vial capacity (FVC) (Griggs et al., 1981; Hough, 1991; also see Ch. 15). Symptoms of incipient respiratory failure include recurrent chest infections, weight loss, early-morning headaches, sweating, disturbed nights and daytime somnolence. A sleep study will confirm nocturnal hypoventilation, and non-invasive respiratory support improves symptoms and can be life saving.
Principles of management
Current therapeutic research is targeted towards finding ways of replacing the abnormal protein. Those individuals with fewer secondary complications of their disorder (e.g. contractures and spinal deformity) will almost certainly derive the greatest benefit if new effective treatments are discovered. Management is based upon the recognition of the patient’s specific needs, together with monitoring for complications of the disorder, e.g. 24-hour cardiac monitoring in EDMD and respiratory monitoring in rigid spine syndrome (RSSI). Corticosteroids have been shown to improve muscle strength in Duchenne muscular dystrophy (DMD), thus prolonging independent walking and are now routinely prescribed from the age of 5 years (Manzur et al., 2008a). But side effects are significant, especially osteoporosis, and must be actively managed.
The muscular dystrophies
The muscular dystrophies are a heterogeneous group of genetically determined disorders associated with progressive degeneration of skeletal muscle. They can be subdivided into a number of different conditions based upon the mode of inheritance, protein, enzyme and/or genetic defect (Tables 10.1). The Xp21 dystrophies or dystrophinopathies are allelic disorders with a wide spectrum of severity with DMD at the most severe end, a group of intermediate patients and BMD at the milder end of the spectrum. X-linked cardiomyopathy (XLDC) is caused by a deletion in the same gene and results in a severe life-limiting cardiomyopathy, but little, if any, muscle weakness. All of these disorders are caused by a defect of the protein dystrophin and are characterized by X-linked inheritance, in which males are affected and females are carriers, although up to 10% of carriers can manifest muscle weakness. Apart from muscle hypertrophy particularly affecting the calf muscles, there is a normal appearance in infancy, although the serum, CK is very high. With advancing age there is progressive muscle weakness and wasting leading to severe physical disability.
Duchenne muscular dystrophy
DMD was first described by Meryon in 1852 and later by Duchenne. It is rapidly progressive and is the most severe of all the muscular dystrophies. The incidence is 1:3500 live male births and there is a prevalence in the population of 1.9–4.8 per 100 000 (Emery, 1991).
Clinical and diagnostic features
By 7–8 years of age, contractures of heel cords and ilio-tibial bands lead to toe walking. Without corticosteroid treatment ambulation is always lost by the 13th birthday and the mean age for loss of ambulation is 9.5 years (Emery & Muntoni, 2003). Prolonged sitting caused by wheelchair dependence leads to the rapid development of flexion contractures of the elbows, hips and knees.
In the early ambulatory phase of DMD, an equinus foot posture is precipitated by relative weakness of the ankle dorsiflexors compared with the better preserved plantar-flexors. Gait analysis has shown that a dynamic equinus is a necessary biomechanical adaptation to maintain knee stability in the presence of gross quadriceps muscle weakness. Forceful action of the ankle plantarflexors provides a torque which opposes knee flexion (Khodadadeh et al., 1986). Thus, contracture of the Achilles tendon, which eventually accompanies disease progression, is secondary to dynamic equinus.
Thus, DMD is a severe life-limiting disease characterized by muscle weakness, contracture, deformity and progressive disability. However, ‘incurable’ is not synonymous with ‘untreatable’. A variety of therapeutic and surgical measures are available that can help to minimize deformity, prolong independent ambulation and maximize functional capabilities. There is evidence that improved management strategies are resulting in increased survival rates (Eagle et al., 2002; Eagle et al., 2007). Corticosteroid treatment results in an increase in muscle strength followed by a slowing down of the dystrophic process (Manzur et al., 2008a). Most boys will die from respiratory or cardiac failure, but the introduction of nocturnal nasal ventilation has improved survival figures, such that the average life expectancy is now 25 years or more with non-invasive ventilation, compared with 19 years without this treatment (Eagle et al., 2002). The principles of successful management are based on an understanding of the natural evolution of patterns of weakness, contracture and deformity, so that intervention can be staged appropriately.
Pathology
Weakness in DMD results from the gradual loss of functional muscle fibres which are replaced by fat and connective tissue due to a lack of dystrophin, a protein encoded by the Xp21 gene, is the primary biochemical defect (Muntoni et al., 2003). Dystrophin is integral within a complex of proteins which stabilizes the integrity of the sarcolemmal membrane, particularly during the stress associated with repeated cycles of contraction and relaxation. Absence of dystrophin in the skeletal and cardiac muscle results in a reduction in permeability of the muscle cell membrane, so allowing excessive quantities of calcium to accumulate within the muscle fibre leading to myofibrillar over-contracture, breakdown of myofibrils and various metabolic disturbances that culminate in muscle fibre degeneration. Dystrophin isoforms are also expressed in Schwann and Purkinge cells found in the brain which is the reason for the high incidence of learning difficulty.
Diagnosis
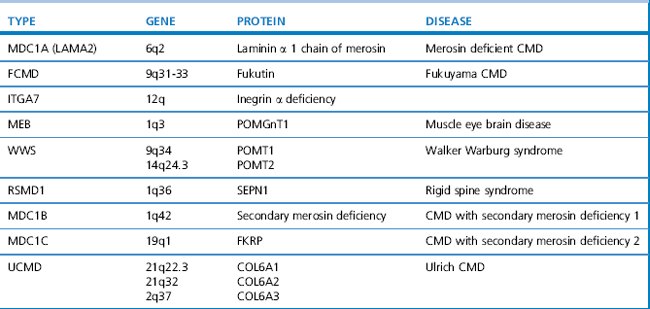
Diagnosis is suspected by finding a raised serum creatine kinase usually in the region of 50–100 times the normal level. A muscle biopsy is necessary to confirm the diagnosis together with DNA analysis. Muscle biopsy can be undertaken as a percutaneous technique using a biopsy needle or as an open procedure, depending on the practice of the investigating unit. Muscle histology demonstrates dystrophic features which include: an increased variation of fibre size, evidence of necrosis with phagocytosis, an increase in central nuclei, hypercontracted eosinophillic hyaline fibres, and an increase in fat and connective tissue (Figure 10.1). Histochemical staining using antibodies to N, C and rod domain epitopes of dystrophin usually show complete absence of the protein, except for occasional revertent fibres (these are fibres which label normally with antibodies to dystrophin; their origin is not understood).
DNA testing from a blood sample will demonstrate a frame-shift deletion within the dystrophin gene in approximately 60% of DMD patients, the rest will result from duplications and nonsense mutations (Mastaglia & Laing, 1996). Confirmation of the diagnosis by DNA testing allows carrier detection and prenatal diagnosis in the affected boy’s mother and female relatives. In some cases DNA analysis is the only test required to confirm the diagnosis of DMD. In BMD the reading frame of the gene is preserved (in-frame). Exceptions to the frame-shift rule occur with some deletions and thus a muscle biopsy is recommended to confirm the diagnosis.
General management
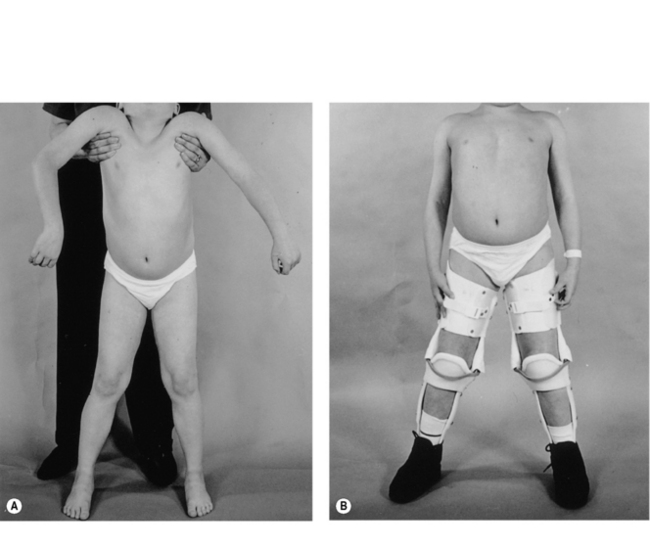
From an early age, tightness and subsequently contractures of the tendo-achilles (TA) develop. Daily passive stretching of the TA and provision of night splints are recommended as soon as any tightening is demonstrated. Later on, when significant pelvic girdle weakness is manifest by a waddling gait, the ilio-tibial bands and hip flexors may start to tighten. Care must be taken to ensure assessment at each outpatient visit with advice to undertake stretching exercises. The priority of physical management is to prolong ambulation for as long as possible, since once ambulation is lost scoliosis and joint contractures develop rapidly. At the point of loss of ambulation, light weight knee-ankle-foot orthoses (KAFOs) can be used to prolong walking (Figure 10.2). This usually involves a minor surgical procedure to percutaneously release any lower limb contractures, usually the TAs, together with an intensive rehabilitation programme. Where there are only moderate contractures of the TAs, serial casting may be an alternative to surgery to allow fitting of the KAFOs (Main et al., 2007). The parents and child must be both strongly motivated and well supported for this form of rehabilitation to be successful.
In recent years, there is growing evidence that treatment with corticosteroids (prednisolone or deflazacort) will stabilize muscle function for some time, therefore delaying the loss of ambulation by up to 2 years (Manzur et al., 2008a). Careful monitoring for side-effects is essential, especially weight gain, which may potentially have a negative effect on function. The potential benefit of longer-term steroid treatment has yet to be evaluated, but some open studies suggest preserved respiratory and cardiac function (Manzur et al., 2008a). Premature loss of ambulation can follow lower limb fractures or ligament strains unless active management, such as internal fixation, is instigated to promote early mobility. Immobilizing any joint beyond the initial painful phase should be actively discouraged.
Once independent ambulation is lost, regular use of a standing frame is recommended to maintain good posture and reduce contracture development. Rigid ankle foot orthoses (AFOs) are recommended for daytime use to maintain a good foot position. Likewise it is essential to ensure that the wheelchair provides good back and neck support (Pope, 2002) and, ideally, the controls of an electric wheel chair should be centrally placed. Swimming and hydrotherapy are particularly useful and enjoyed by the boys who find they can move more easily in the water.
A rapidly progressive scoliosis requiring surgical stabilization will develop in up to 95% of boys. There is, however, a reduced incidence and severity of scoliosis in glucocorticosteroid-treated boys (Biggar et al., 2006) which is likely to be due to prolongation of walking and increase in trunk muscle strength. Posterior spinal fusion is highly successful in correcting the deformity, and in improving posture and the quality of life for both patient and carer (Mehdian et al., 1989). The timing of operation is crucial since a decline in vital capacity occurs at the same time as the development of scoliosis. To avoid undue anaesthetic risks the procedure must be undertaken when the vital capacity is greater than 30% of predicted height (Manzur et al., 2008b). Thus surgical correction is usually recommended when the degree of spinal curvature is still relatively mild.
Management of restricted participation
Restricted participation has replaced the term ‘handicap’ in the revised International Classification of Functioning, Disability and Health by the World Health Organization (WHO ICF, 2001; see Ch. 11) and its management must take into account psychosocial issues, mobility and education. Providing an electric wheelchair is essential to providing a degree of independence, but this requires appropriate home adaptations including: widened doors and indoor/outdoor wheelchair access. A hoist is required for lifting in and out of the wheelchair, and the child will require a ground floor bedroom and bathroom with shower and adapted toilet. An electric bed enables the child to alter his position and saves the parents many sleepless nights. An adapted motor vehicle is required for transport to enable the child to drive his own wheelchair in and out of the vehicle.
Respiratory problems
With advancing age, respiratory impairment becomes inevitable and, if not recognized early, is an important cause of unpleasant symptoms or death. Characteristically there is a restrictive defect, with a reduction in total lung capacity caused by a combination of diaphragmatic and intercostal muscle weakness. Chest wall stiffness, recurrent aspiration and an inability to cough effectively compound the respiratory insufficiency leading to an increased frequency of chest infections (Smith et al., 1991). The forced vital capacity is a reliable measure of respiratory function, provided the boy is able to undertake a good technique (in some boys with learning difficulty this may be a problem) (Griggs et al., 1981). The forced vital capacity, when corrected for height, plateaus and then falls progressively on average between 12 and 14 years of age. Once the vital capacity falls below 1 L, in a boy who has reached skeletal maturity, the average life expectancy without treatment is 3 years (Phillips et al., 2001).
Sleep-related respiratory abnormalities play a major role in ventilatory failure, resulting in symptoms of hypercapnia which include: early-morning headache, nausea and sweating, daytime somnolence and a loss of respiratory drive (resulting in rapid deterioration into coma if a high concentration of oxygen is administered). Chronic nocturnal hypoxaemia leads to cor pulmonale (right heart failure), the ECG may show evidence of pulmonary hypertension and right heart strain (Carroll et al., 1991). Once the vital capacity falls below 40% sleep studies should be undertaken at regular intervals to monitor for nocturnal hypoventilation. Treatment by non-invasive nasal ventilation is effective in alleviating symptoms and prolongs survival (Eagle et al., 2002; Jeppesen et al., 2003; Simonds, 2000). There is recent evidence that the cumulative effect of both spinal surgery and nocturnal ventilation further improves survival (Eagle et al., 2007).
Cardiac problems
Post-mortem studies show that all boys with DMD have evidence of cardiomyopathy by 18 years of age. In practice, however, symptomatic cardiomyopathy is less common than might be expected. It has been assumed the sedentary lifestyle of these boys contributes to the lack of symptoms (Hunsaker et al., 1982). Abnormalities of the electrocardiogram are evident from an early age and will be present in all boys by 18 years of age (Nigro et al., 1990); the most common abnormality is a resting tachycardia, which is almost universal. Cardiac arrhythmias occur and may be a cause of early sudden death. When congestive cardiac failure does occur the progression is rapid and relentless. Monitoring with cardiac echo is recommended once ambulation is lost. Early treatment with ACE (angiotensin-converting enzyme) inhibitors has been shown to be beneficial, although the results need to be confirmed (Duboc et al., 2007).
The final illness
Close monitoring and liaison with the general practitioner and palliative care services are paramount. Children’s Respite Hospices play a vital role in supporting families and boys during this difficult time. Patient support groups, such as The Muscular Dystrophy Campaign, frequently fund care workers to support families (see Appendix ‘Associations and Support Groups’).
Becker muscular dystrophy
Becker muscular dystrophy (BMD) is allelic to DMD, but has a milder phenotype (see Table 10.1) and has a prevalence of 1 in 30 000. It is caused by a partial deficiency of the protein dystrophin (Karpati et al., 2001).
Clinical and diagnostic features
BMD has a wide spectrum of severity; at the severest end ambulation may be lost by 16 years of age compared with the mildest form which presents with non-progressive cramps and myoglobinuria (Gospe et al., 1989). Distribution of weakness is similar to DMD, but progression of the disease is much slower and contractures are often less severe than in DMD. As with DMD, muscle hypertrophy, especially of the calves, occurs. Up to 40% of patients will lose ambulation, and prolonging ambulation with long-leg calipers is more difficult than in DMD because of adult height.
In some patients, as with DMD, an unexpected malignant hyperthermia reaction following general anaesthesia may be the first manifestation of the disease. Cardiomyopathy is common and more likely to be symptomatic than in DMD. ECG and echocardiogram abnormalities may be evident in up to 50% of cases (Steare et al., 1992) and many patients have successfully undergone cardiac transplantation (Quinlivan & Dubowitz, 1992).
General management
The management of BMD involves prevention of contractures and prolonging ambulation as with DMD. An active approach to management using low-intensity aerobic exercise has been shown to safely improve fitness and strength in individuals with mild BMD (Sveen et al., 2008). Home adaptations are essential in promoting independence and the patient may require support to continue working in an adapted environment. For those patients who are wheelchair dependent, regular standing, and preventing excessive weight gain and constipation are important. Prevention of respiratory infections by vaccination against influenza and pneumococcus, together with prompt antibiotic treatment of infection are important. Monitoring of respiratory function and overnight oximetry for sleep hypoxaemia are necessary. Symptoms of chronic ventilatory failure should be managed with non-invasive ventilation as for the DMD group. Regular cardiac monitoring with yearly ECGs and cardiac ECHOs every 2 or 3 years is necessary. Early intervention with ACE inhibitors for ventricular dysfunction may be helpful, but as yet has to be fully evaluated. If cardiac symptoms fail to respond to medical treatment, assessment for cardiac transplantation is warranted (Quinlivan & Dubowitz, 1992).
Emery–Dreifuss muscular dystrophy
This is a rare but clinically distinct form of MD. Two modes of inheritance exist. Firstly, a sex-linked form (EDMD) in which mutations in the Emerin gene located at Xq28 lead to a complete absence of the nuclear envelope protein Emerin (Mastaglia & Laing, 1996). Secondly, there is an autosomal dominant form (ADEDMD) in which the defective gene is at 1q11-q23 encoding for another nuclear envelope protein; Lamin A/C (LMNA) (Bonne et al., 1999).
Clinical and diagnostic features
The main feature of the nuclear envelope muscular dystrophies is the predominance of joint contractures and muscle weakness occurring in a scapulo-peroneal distribution. Contractures predominantly affect the neck and spine, elbows and tendo-achilles. Disturbances of cardiac conduction are universal by the second or third decades, and may result in sudden death unless detected and cardiac pacing instituted bb0025(Bialer et al., 1991). Thus, regular ECG and 24-hour ECG monitoring are essential. Muscle weakness is slowly progressive, however many patients retain some ambulation throughout adult life.





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


