Myotonic dystrophy is the most common myotonic disorder (Table 31-1). There are at least two genetically distinct forms of myotonic dystrophy: Dystrophica myotonia type 1 (DM1) and dystrophica myotonia type 2 (DM2), the later of which is also known as proximal myotonic myopathy (PROMM).
|
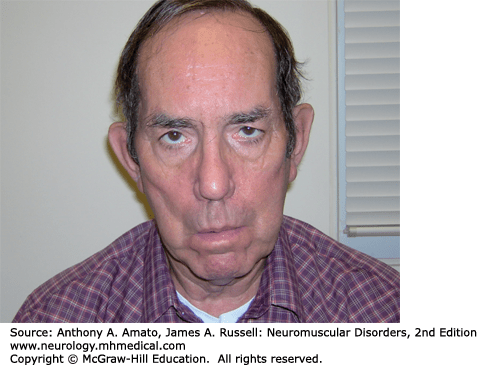
DM1 in an autosomal dominant manner with a prevalence of 13.5 per 100,000.1–4 DM1 can present at any age, including infancy. Limb weakness begins distally in the extremities and can progress slowly to affect proximal muscles. Wrist flexors are often weaker than wrist extensors. The neck flexors, including the sternocleidomastoids, are also affected early. Atrophy and weakness of temporalis and other facial muscles as well as the jaw muscles giving rise to the characteristic “hatchet face” appearance (Fig. 31-1). Ptosis is often evident. Some patients develop dysarthria and dysphagia due to pharyngeal and lingual muscles involvement.
Many patients do not complain or are not aware of their myotonia, although it is usually readily apparent on examination, particularly in the hands. Delayed relaxation of the fingers is seen following a forceful hand grip (action myotonia). The myotonia is lessened with repeated muscle contractions, a so-called warm-up phenomenon. Percussion of muscle groups, in particular of the thenar eminence or finger extensors also gives rise to delayed relaxation (percussion myotonia). Muscle reflexes are diminished, but sensory testing is normal.5 Adult patients with DM1 may have a mild reduction in cognitive abilities, while severe mental retardation is associated with congenital myotonic dystrophy.6,7
Congenital myotonic dystrophy is much more severe than adult-onset DM1. Affected infants are invariably born to mothers with myotonic dystrophy.8,9 It is important to examine mothers of floppy infants, as they may not even be aware that they have the disorder. Pregnancy may be complicated by polyhydramnios and diminished fetal movements. Infants with congenital myotonic dystrophy have severe generalized weakness and hypotonia and may also have arthrogryposis. Clinical myotonia is not apparent in the neonatal period and may not be noticeable until about 5 years of age. However, myotonic discharges can be appreciated on electromyography (EMG) before the appearance of clinical myotonia. Many infants require ventilatory assistance due to ventilatory insufficiency. The mortality rate in infancy is approximately 25%. Severe psychomotor abnormalities affect 75% of surviving children. Most will have some degree of mental retardation. Life expectancy is greatly reduced in DM1 patients, particularly those with early onset of the disease and significant proximal, in addition to distal, weakness.10,11
DM1 is a systemic disorder affecting the gastrointestinal tract, the uterus, ventilatory muscles, cardiac muscle, the lens, and the endocrine system.12 In addition to dysphagia, reduced gastrointestinal motility can lead to chronic pseudo-obstruction.13,14 Alveolar hypoventilation can arise from involvement of the diaphragm and intercostal muscles. It is more severe in congenital myotonic dystrophy and may lead to ventilatory failure, but this certainly occurs in later onset cases as well.9 It is unclear if decreased central drive contributes to hypoventilation.15,16 Nonetheless, many patients develop symptoms suggestive of sleep apnea: frequent nocturnal arousals, excessive daytime hypersomnolence, and morning headaches. Pulmonary hypertension can develop and may lead to cor pulmonale.
Cardiac abnormalities are common with approximately 90% of patients having conduction defects on electrocardiograms (EKGs).17 Sudden cardiac death secondary to arrhythmia is well documented. However, the severity of the cardiomyopathy does not necessarily correlate with the severity of skeletal muscle weakness. The size of the mutation (discussed in Pathogenesis section) and the severity of the skeletal muscle weakness do not correlate with the occurrence of cardiac conduction abnormalities or sudden death.18 It seems that risk of sudden death increases with duration of disease and age, and that risk is higher in male patients.18
Neurobehavioral abnormalities are common in patients with DM1.19,20 Neuropsychological testing demonstrates elements of obsessive–compulsive, passive–aggressive, dependent, and avoidant personality traits in many patients. Apathy and depression are also frequent. Cognitive impairment, particularly in memory and spatial orientation, may be demonstrated. The neuropsychological deficits appear to correlate with brain single photon emission computed tomography, which shows frontal and parieto-occipital hypoperfusion.20
Other systemic manifestations include posterior subscapular cataracts, frontal balding, testicular atrophy and impotence in men, and a high rate of fetal loss and complications of pregnancy in women. Hyperinsulinemia is common following glucose tolerance tests, however, the frequency of overt diabetes mellitus is not increased.21
Some epidemiological studies have reported an increased risk of cancer in patients with DM1. In a study of Swedish and Danish populations, the risk of malignancy was double that of the general population.22 Specifically, they observed an increased risk of endometrial, ovarian, colon, and brain cancer. In a study from the Mayo Clinic, there was an increased risk of thyroid cancer and choroidal melanoma, as well as perhaps testicular and prostate cancer.23 However, they found no increased risk of endometrial, ovarian, breast, colorectal, lung, renal, bladder, or brain cancers.
Serum creatine kinase (CK) may be normal or mildly increased. Motor and sensory nerve conduction studies (NCS) are usually normal. EMG demonstrates myotonic discharges (Fig. 31-2). It is important to sample multiple muscles as myotonic discharges are not necessarily appreciated in every muscle studied.24 Facial and intrinsic hand muscles are the most commonly affected. In congenital myotonic dystrophy, electrical myotonia may be observed as early as 5 days to 3 weeks following birth and increases with age.25,26 Fibrillation potentials, positive sharp waves, and myopathic motor unit action potentials (MUAPs) may also be seen but they can be obscured by the myotonic discharges.

Muscle biopsies demonstrate an increased number of internalized nuclei in the muscle fibers (Fig. 31-3). Type 1 predominance and atrophy are very common. In addition, hypertrophic type 2 fibers, ring fibers, small angulated fibers, atrophic fibers with pyknotic nuclear clumps, and sarcoplasmic masses are also frequently observed. In contrast to other muscular dystrophies, necrotic fibers and increased connective tissue are less conspicuous. Autopsy studies of the brain demonstrate neurofibrillary degeneration with abnormal tau expression.27

DM1 is caused by an expansion of unstable polymorphic cytosine–thymine–guanine (CTG) trinucleotide repeats in the 3′ untranslated region of the myotonin protein kinase gene, (DMPK), that is located on chromosome 19q13.2.12,28–35 This CTG repeat is copied in the gene up to 27 times in normals, but 50 to more than 4,000 copies are found in DM1 patients. The severity of the myopathy directly correlates with the size of the CTG repeat, which is unstable. The mutation size usually expands from one generation to the next, which accounts for the anticipation phenomena (i.e., the earlier presentation and/or more severe disease in each generation). More marked expansion of the CTG repeat usually occurs in children of mothers with DM1, which explains the severe phenotype of congenital myotonic dystrophy.
It is not the abnormal expression of myotonin protein kinase itself that is responsible for the disorder. Rather, DM1 seems to be a consequence of nuclear retention of mutant mRNA containing expanded CTG repeats, rather than a specific lack or gain of function of the DMPK protein. Indeed, the myopathy and other systemic features appear to be due to a toxic gain of function of the mutant mRNA.36
The transcribed mRNA with expanded CTG (DM1) accumulates as abnormal focal collections in the nucleus that cannot be transported to the cytoplasm, where RNA translation into protein takes place.37–40 Aggregates of mutated mRNA are directly toxic to cells by sequestering RNA-binding proteins (such as muscleblind proteins), which in turn, lead to abnormal splicing of pre-mRNA from various target genes (e.g., chloride ion channel, insulin receptor, tau protein, cardiac troponin, ryanodine receptor, and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase).37–39,41–45 Therefore, there is abnormal translation of the RNAs into functional proteins, and this explains the multiple organ/systemic manifestations of DM1. Other studies have shown that mutant RNA binds and sequesters transcription factors with up to 90% depletion of selected transcription factors from active chromatin.46 This leads to reduced expression of a variety of genes, including the chloride ion channel (CIC-1), which is also mutated in myotonia congenita and is the likely origin of the myotonic discharges that occur in both disorders.
There are no medical therapies that clearly improve muscle strength. A small pilot study of dehydroepiandrosterone sulfate (DHEAS) in 11 patients with DM1 seemed to be beneficial in a few patients, but larger controlled trials are necessary before commenting on the possible efficacy.47 Small trials of creatine monohydrate in DM1 failed to demonstrate efficacy.48 A recent study of recombinant human insulin-like growth factor 1 (rhIGF-1) complexed with IGF-binding protein 3 (rhIGF-1/rhIGFBP-3) in patients with DM1 reported that drug was associated with increased lean body mass and improvement in metabolism, but not increased muscle strength or function.49
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


