Fig. 26.1
Hypocretin deficiency in narcoleptic subjects. a CSF hypocretin-1 levels are undetectably low in most narcoleptic subjects (84.2 %). Note that two HLA DQB1*0602-negative and one familial case have normal or high CSF hypocretin levels. b Prepro-hypocretin transcripts are detected in the hypothalamus of control (b) but not in narcoleptic subjects (a). Melanin-concentrating hormone ( MCH) transcripts are detected in the same region in both control (d) and narcoleptic (c) sections. c Colocalization of IGFBP3 in HCRT cells in control and narcolepsy human brain. Upper panel: e Distribution of hypocretin cells and fibers in the perifornical area of human hypothalamus. f In control brains, HCRT cells and fibers were densely stained by an anti-HCRT monoclonal antibody (red fluorescence: VectorRed), while in narcolepsy brains, staining was markedly reduced. Lower panel: HCRT immunoreactivity (g: red fluorescence) and IGFBP3 immunoreactivity (h: green fluorescence; Q-dot525) and a composite picture (i) arrows indicate HCRT cells colocalized with IGFBP3). Note: nonneuronal autofluorescent elements. f and fx, fornix. Scale bar represents 10 mm (a–d), 500 mm in (e and f), 100 mm in g, h, and i (from [78] and [81]). CSF cerebrospinal fluid, HLA human leukocyte antigen, HCRT hypocretin, IGFBP3 insulin-like growth factor-binding protein 3
As a result of these findings, a diagnostic test for narcolepsy based on clinical measurement of CSF hypocretin-1 levels for detecting hypocretin ligand deficiency is now available [1]. Whereas CSF hypocretin-1 concentrations above 200 pg/ml almost always occur in controls and patients with other sleep and neurological disorders, concentrations below 110 pg/ml are 94 % predictive of narcolepsy with cataplexy [79] (Fig. 26.2). As this represents a more specific assessment than the multiple sleep latency test (MSLT), CFS hypocretin-1 levels below 110 pg/ml are indicated in the International Classification of Sleep Disorders (ICSD)-3 as diagnostic of narcolepsy with cataplexy [1].
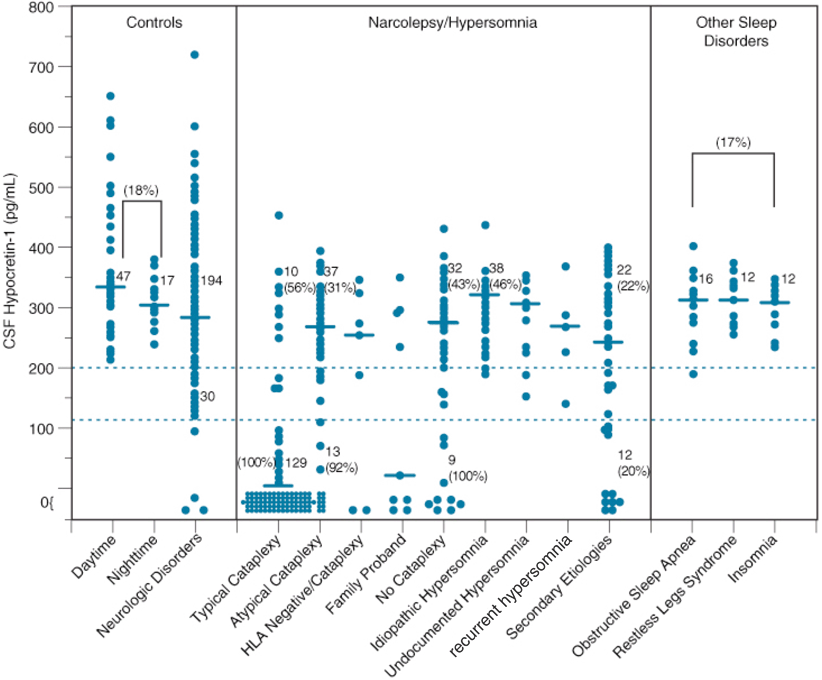
Fig. 26.2
CSF hypocretin-1 levels in individuals across various control and sleep disorders. Each point represents the crude concentration of hypocretin-1 in a single person. The cutoffs for normal (> 200 pg/mL) and low (< 110 pg/mL) hypocretin-1 concentrations are shown. Also noted is the total number of subjects in each range, and the percentage human leukocyte antigen (HLA)-DQB1*0602 positivity for a given group in a given range is parenthetically noted for certain disorders. Note that control carrier frequencies for DQB1*0602 are 17–22 % in healthy control subjects and secondary narcolepsy, consistent with control values reported in whites (see Table 64.3). In other patient groups, values are higher, with almost all hypocretin-deficient narcolepsy being HLA DQB1*0602 positive. The median value in each group is shown as a horizontal bar. (Updated from previously published data [79])
Moreover, separate coding of “narcolepsy with cataplexy” (type 1) and “narcolepsy without cataplexy” (type 2) in the ICSD-3 underscores how discovery of specific diagnostic criteria now informs our understanding of narcolepsy’s nosology; narcolepsy with cataplexy, as indicated by low CSF hypocretin-1, appears etiologically homogeneous and distinct from most patients with narcolepsy without cataplexy, exhibiting normal hypocretin-1 levels [79]. Further, the potential of hypocretin receptor agonists (or cell transplantation) in narcolepsy treatment is currently being explored, and CSF hypocretin-1 measures may be useful in identifying appropriate patients as candidates for a novel therapeutic option, namely hypocretin replacement therapy.
Soon after the discovery of human hypocretin deficiency , researchers identified specific substances and genes, such as dynorphin and neuronal activity-regulated pentraxin (NARP) [80] and most recently, insulin-like growth factor-binding protein 3 (IGFBP3) [81], which colocalizes in neurons containing hypocretin. These findings underscored selective hypocretin cell death as the cause of hypocretin deficiency (as opposed to transcription/biosynthesis or hypocretin peptide processing problems), because these substances are also deficient in postmortem brain HLA of hypocretin-deficient narcoleptic patients [80, 81]. Further, these findings, in view of the generally late onsets of sporadic narcolepsy compared with those of familial cases, suggest that postnatal cell death of hypocretin neurons constitutes the major pathophysiological process in human narcolepsy with cataplexy.
A large kindred with familial narcolepsy (12 affected members) has been reported in Spain [82]. Affected members do not exhibit any symptoms suggesting symptomatic cases of narcolepsy and were diagnosed as familial idiopathic narcolepsy–cataplexy . The family includes a pair of dizygotic twins concordant for narcolepsy–cataplexy in the third generation; the distribution of the disorder indicates an autosomal-dominant transmission of the disease-causing gene. Hor et al. recently performed linkage analysis and sequenced coding regions of the genome (exome sequencing) of three affected members with narcolepsy and cataplexy and had identified a missense mutation in the second exon of myelin oligodendrocyte glycoprotein (MOG) [82]. A c.398 C > G mutation was present in all affected family members but absent in unaffected members and 775 unrelated control subjects [82]. Affected members were hypocretin deficient, but association with HLA DQB1*0602 was not observed [82]. The mutation may secondarily induce hypocretin deficiency with or without immune-mediated mechanisms . MOG has recently been linked to various neuropsychiatric disorders and is considered as a key autoantigen in multiple sclerosis (MS) and in its animal model, experimental autoimmune encephalitis [83]; thus autoimmune mechanisms may also be involved in these cases. However, even if autoimmune mechanisms are involved in these cases, it is possible that the primary target for the immune attack is not the hypocretin system. These results also suggest the heterogeneity of etiology of idiopathic narcolepsy–cataplexy .
How Does Hypocretin Ligand Deficiency Cause the Narcolepsy Phenotype?
Since hypocretin deficiency is a major pathophysiological mechanism for narcolepsy–cataplexy, how the hypocretin ligand deficiency can cause the narcolepsy phenotype is discussed.
Hypocretin/Orexin System and Sleep Regulation
Hypocretins/orexins (hypocretin-1 and hypocretin-2/orexin A and orexin B) are cleaved from a precursor prepro-hypocretin (prepro-orexin) peptide [72, 73, 84]) (Fig. 26.3). Hypocretin-1 with 33 residues contains four cysteine residues forming two disulfide bonds. Hypocretin-2 consists of 28 amino acids and shares similar sequence homology especially at the C-terminal side but has no disulfide bonds (a linear peptide) [72]. There are two G-protein-coupled hypocretin receptors, Hcrtr 1 and Hcrtr 2, also called orexin receptor 1 and 2 (OX1R and OX2R), and distinct distribution of these receptors in the brain is known. Hcrtr 1 is abundant in the LC while Hcrtr 2 is found in the TMN and basal forebrain (Fig. 26.3). Both receptor types are found in the midbrain raphe nuclei and mesopontine reticular formation [4].
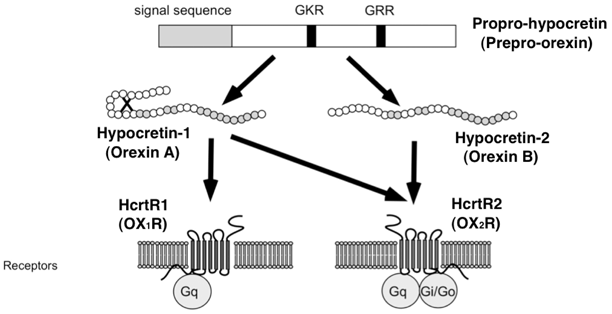
Fig. 26.3
a Structures of mature hypocretin-1 (orexin A) and hypocretin-2 (orexin B) peptides. b Schematic representation of the hypocretin (orexin) system. c Projections of hypocretin neurons in the rat brain and relative abundance of hypocretin receptor 1 and 2. a The topology of the two intrachain disulfide bonds in orexin A is indicated in the above sequence. Amino acid identities are indicated by shaded areas. b The actions of hypocretins are mediated via two G-protein-coupled receptors named hypocretin receptor 1 ( Hcrtr 1) and hypocretin receptor 2 ( Hcrtr 2), also known as orexin-1 ( OX 1 R) and orexin-2 ( OX 2 R) receptors, respectively. Hcrtr 1 is selective for hypocretin-1, whereas Hcrtr 2 is nonselective for both hypocretin-1 and hypocretin-2. Hcrtr 1 is coupled exclusively to the G q subclass of heterotrimeric G proteins, whereas in vitro experiments suggest that Hcrtr 2 couples with G i/o, and/or G q. (adapted from Sakurai (2002). c Hypocretin-containing neurons project to these previously identified monoaminergic and cholinergic and cholinoceptive regions where hypocretin receptors are enriched. The relative abundance of Hcrtr 1 versus Hcrtr 2 in each brain structure was indicated in parenthesis (data from Marcus et al. 2001). Impairments of hypocretin input may thus result in cholinergic and monoaminergic imbalance and generation of narcoleptic symptoms. Most drugs currently used for the treatment of narcolepsy enhance monoaminergic neurotransmission and adjust these symptoms. VTA ventral tegmental area, SN substantia nigra, LC locus coeruleus, LDT laterodorsal tegmental nucleus, PPT pedunculopontine tegmental nucleus, RF reticular formation, BF basal forebrain, VLPO ventrolateral preoptic nucleus, LHA lateral hypothalamic area, TMN tuberomammillary nucleus, DR dorsal raphe, Ach acetylcholine, Glu glutamate, GABA γ-aminobutyric acid, HI histamine, DA dopamine, NA noradrenalin, 5-HT serotonin
Hypocretins-1 and -2 are produced exclusively by a well-defined group of neurons localized in the lateral hypothalamus. The neurons project to the olfactory bulb, cerebral cortex, thalamus , hypothalamus, and brainstem, particularly the LC, raphe nucleus, and to the cholinergic nuclei (the laterodorsal tegmental and pedunculopontine tegmental nuclei) and cholinoceptive sites (such as pontine reticular formation) [74, 84]. All of these projection sites are thought to be important for sleep regulation.
A series of recent studies have now shown that the hypocretin system is a major excitatory system that affects the activity of monoaminergic (DA, NE, 5-HT, and histamine) and cholinergic systems with major effects on vigilance states [84, 85]. It is thus likely that a deficiency in hypocretin neurotransmission induces an imbalance between these classical neurotransmitter systems, with primary effects on sleep-state organization and vigilance.
Many measurable activities (brain and body) and compounds manifest rhythmic fluctuations over a 24-h period. Whether or not hypocretin tone changes with zeitgeber time was assessed by measuring extracellular hypocretin-1 levels in the rat brain CSF across 24-h periods, using in vivo dialysis [86]. The results demonstrate the involvement of a slow diurnal pattern of hypocretin neurotransmission regulation (as in the homeostatic and/or circadian regulation of sleep). Hypocretin levels increase during the active periods and are highest at the end of the active period, and the levels decline with the onset of sleep. Furthermore, sleep deprivation increases hypocretin levels [86].
Recent electrophysiological studies have shown that hypocretin neurons are active during wakefulness and reduce the activity during slow-wave sleep [87]. The neuronal activity during REM sleep is the lowest, but intermittent increases in the activity associated with body movements or phasic REM activity are observed [87]. In addition to this short-term change, the results of microdialysis experiments also suggest that basic hypocretin neurotransmission fluctuates across the 24-h period and slowly builds up toward the end of the active period. Adrenergic LC neurons are typical wake-active neurons involved in vigilance control , and it has been recently demonstrated that basic firing activity of wake-active LC neurons also significantly fluctuates across various circadian times [88].
Several acute manipulations such as exercise, low glucose utilization in the brain, and forced wakefulness increase hypocretin levels [85, 86]. It is therefore hypothesized that a build up/acute increase of hypocretin levels may counteract homeostatic sleep propensity that typically increases during the daytime and during forced wakefulness [89].
Hypocretin/Orexin Deficiency and Narcoleptic Phenotype
Human studies have demonstrated that the occurrence of cataplexy is closely associated with hypocretin deficiency [79]. Furthermore, the hypocretin deficiency was already observed at very early stages of the disease (just after the onset of EDS) , even before the occurrences of clear cataplexy. Occurrences of cataplexy are rare in acute symptomatic cases of EDS associated with a significant hypocretin deficiency (see [3]); therefore, it appears that a chronic and selective deficit of hypocretin neurotransmission may be required for the occurrence of cataplexy. The possibility of involvement of a secondary neurochemical change for the occurrence of cataplexy still cannot be ruled out. If some of these changes are irreversible, hypocretin supplement therapy may only have limited effects on cataplexy.
Sleepiness in narcolepsy is most likely due to the difficulty in maintaining wakefulness as normal subjects do. The sleep pattern of narcoleptic subjects is also fragmented; they exhibit insomnia (frequent wakening) at night. This fragmentation occurs across 24 h, thus, the loss of hypocretin signaling is likely to play a role in this vigilance stage stability (see [90]), but other mechanism may also be involved in EDS in narcoleptic subjects. One of the most important characteristics of EDS in narcolepsy is that sleepiness is reduced and patients feel refreshed after a short nap, but this does not last long as they become sleepy within a short period of time. Hypocretin-1 levels in the extracellular space and in the CSF of rats significantly fluctuate across 24 h and build up toward the end of the active periods [89] . Several manipulations (such as sleep deprivation, exercise, and long-term food deprivation) are also known to increase the hypocretin tonus [86, 89]. Thus, the lack of this hypocretin build up (or increase) caused by circadian time and by various alerting stimulations may also play a role for EDS associated with hypocretin-deficient narcolepsy.
Mechanisms for cataplexy and REM sleep abnormalities associated with impaired hypocretin neurotransmission have been studied. Hypocretin strongly inhibits REM sleep and activates brainstem REM-off LC and raphe neurons and REM-on cholinergic neurons as well as local GABAnergic neurons. Therefore, disfacilitation of REM-off monoaminergic neurons and stimulation of REM-on cholinergic neurons mediated through disfacilitation of inhibitory GABAnergic inert neurons associated with impaired hypocretin neurotransmission are proposed for abnormal manifestations of REM sleep.
Considerations for the Pathophysiology of Narcolepsy with Normal Hypocretin Levels
There are debates about the pathophysiology of narcolepsy with normal hypocretin levels. Over 90 % patients with narcolepsy without cataplexy show normal CSF hypocretin levels, yet they show apparent REM sleep abnormalities (i.e., SOREMS). Furthermore, even if the strict criteria for narcolepsy–cataplexy are applied, up to 10 % of patients with narcolepsy–cataplexy show normal CSF hypocretin levels. Considering the fact that occurrence of cataplexy is tightly associated with hypocretin deficiency, impaired hypocretin neurotransmission is still likely involved in narcolepsy–cataplexy with normal CSF hypocretin levels. Conceptually, there are two possibilities to explain these mechanisms: (1) specific impairment of hypocretin receptor and their downstream pathway and (2) partial/localized loss of hypocretin ligand (yet exhibit normal CSF levels). A good example for (1) is Hcrtr-2-mutated narcoleptic dogs; they exhibit normal CSF hypocretin-1 levels [91] while having a full-blown narcolepsy. Thannickal et al. recently reported one narcolepsy without cataplexy patient (HLA typing was unknown) who had an overall loss of 33 % of hypocretin cells compared to normal, with maximal cell loss in the posterior hypothalamus [92]. This result favors the second hypothesis, but studies with more cases are needed.
Idiopathic Hypersomnia: A Hypocretin Nondeficient Primary Hypersomnia
With the clear definition of narcolepsy (cataplexy and dissociated manifestations of REM sleep), it became apparent that some patients with hypersomnia suffer from a different disorder. Bedrich Roth was the first in the late 1950s and early 1960s to describe a syndrome characterized by EDS , prolonged sleep, and sleep drunkenness, and by the absence of “sleep attacks,” cataplexy, sleep paralysis, and hallucinations. The terms “independent sleep drunkenness” and “hypersomnia with sleep drunkenness” were initially suggested [93], but now this syndrome is categorized as idiopathic hypersomnia (1). Idiopathic hypersomnia should therefore not be considered synonymous with hypersomnia of unknown origin.
In the absence of systematic studies, the prevalence of idiopathic hypersomnia is unknown. Nosologic uncertainty causes difficulty in determining the epidemiology of the disorder. Recent reports from large sleep centers reported the ratio of idiopathic hypersomnia to narcolepsy to be 1:10. [94]. The age of onset of symptoms varies, but it is frequently between 10 and 30 years. The condition usually develops progressively over several weeks or months. Once established, symptoms are generally stable and long lasting, but spontaneous improvement in EDS may be observed in up to one quarter of patients [94].
The pathogenesis of idiopathic hypersomnia is unknown. Hypersomnia usually starts insidiously. Occasionally, EDS is first experienced after transient insomnia, abrupt changes in sleep–wake habits, overexertion, general anesthesia, viral illness, or mild head trauma [94]. Despite reports of an increase in HLA DQ1,11 DR5 and Cw2, and DQ3, and decrease in Cw3, no consistent findings have emerged [94].
Nosological and Diagnostic Considerations of Major Primary Hypersomnias
Narcolepsy–cataplexy , narcolepsy without cataplexy, and idiopathic hypersomnia are diagnosed mostly by sleep phenotypes, especially by the occurrences of cataplexy and SOREMPS (Fig. 26.4; ICSD-3). Discovery of hypocretin deficiency in narcolepsy–cataplexy was not only a breakthrough but also brought a new nosological and diagnostic uncertainty of the primary hypersomnias. Up to 10 % of patients with narcolepsy–cataplexy show normal CSF hypocretin-1 levels (Fig. 26.4). As discussed above, altered hypocretin neurotransmissions may still be involved in some of these cases. However, up to 10 % of patients with narcolepsy without cataplexy instead show low CSF hypocretin-1 levels, suggesting a substantial pathophysiological overlap between narcolepsy–cataplexy and narcolepsy without cataplexy, and the hypocretin-deficient status (measured in CSF) does not completely separate these two disease conditions (Fig. 26.4). Similarly, concerns about the nosology of narcolepsy without cataplexy and idiopathic hypersomnia should also be addressed. Since patients with typical cases of idiopathic hypersomnia exhibit unique symptomatology, such as long hours of sleep, no refreshment from naps, and generally resistance to stimulant medications, the pathophysiology of idiopathic hypersomnia may be distinct from that of narcolepsy without cataplexy. However, current diagnostic criteria are not specific enough to diagnose these disorders, especially since the test–retest reliability of numbers of SOREMS during MSLT has not been systematically evaluated.
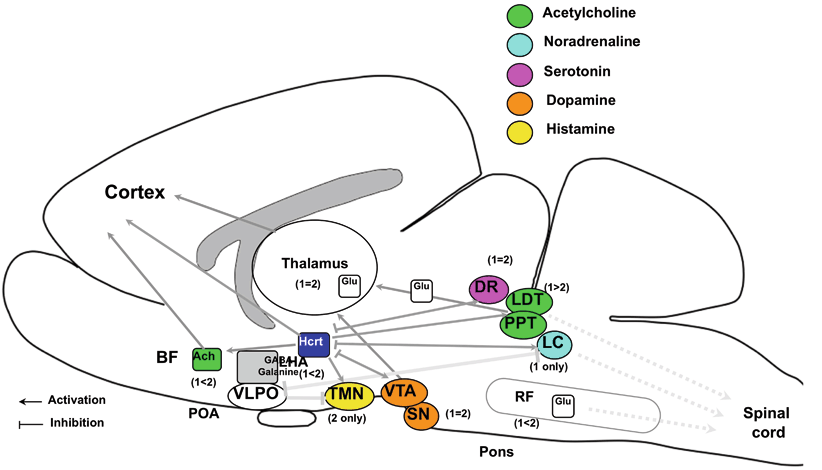
Fig. 26.4
Nosological and diagnostic considerations of major primary hypersomnias. Narcolepsy–cataplexy, narcolepsy without cataplexy, and idiopathic hypersomnia are diagnosed by the occurrences of cataplexy and SOREMPS. Pathophysiology-based marker and low CSF hypocretin levels are included in the ICSD-3 for the positive diagnosis for narcolepsy–cataplexy. However, up to 10 % of patients with narcolepsy–cataplexy show normal CSF hypocretin levels. In contrast, up to 10 % of patients with narcolepsy without cataplexy show low CSF hypocretin-1 levels. These results suggest a substantial pathophysiological overlap between narcolepsy–cataplexy and narcolepsy without cataplexy. Similarly, a substantial overlap likely exists between narcolepsy without cataplexy and idiopathic hypersomnia, as these disorders are diagnosed by the occurrences of SOREMS (two or more). However, the test–retest reliability of detecting number of SOREMS in these conditions has not been systematically evaluated
CSF Histamine and GABAA Receptor Modulator in Narcolepsy and Hypersomnia
Although pathophysiology of hypocretin nondeficient hypersomnia is largely unknown, neurochemical changes in these disease conditions, namely reduced CSF histamine contents and increased activity of GABAA receptor modulator in the CSF, have been reported recently by two groups [96–98].
Histamine is one of these wake-active monoamines [99], and low CSF histamine levels are also found in narcolepsy with hypocretin deficiency [96, 97]. Since hypocretin neurons project and excite histamine neurons in the posterior hypothalamus, it is conceivable that impaired histamine neurotransmission may mediate sleep abnormalities in hypocretin-deficient narcolepsy. However, low CSF histamine levels were also observed in narcolepsy with normal hypocretin levels, and in idiopathic hypersomnia , decreased histamine neurotransmission may be involved in a broader category of EDS than in hypocretin-deficient narcolepsy [97]. Since CSF histamine levels are normalized in EDS patients treated with wake-promoting compounds, low CSF histamine levels may be a new state marker for the hypersomnia of central origin, and functional significances of this finding should further be studied further [97].
Ryer et al. recently reported that activities of substance in CSF that augments inhibitory GABA signaling are enhanced in hypersomnia [98]. The authors demonstrated that in the presence of GABA (10 µM), CSF can stimulate GABAA receptor function in vitro (measures of GABAAR-mediated chloride currents in recombinant pentameric human GABAAR-expressed cultured cells). Interestingly, stimulations of GABAA receptor function by CSF from hypersomnolent patients (idiopathic hypersomnia with and without long sleep , long sleepers and narcolepsy without cataplexy) are significantly enhanced compared to those by CSF from control subjects (84.0 vs. 35.8 %) [98]. This bioactive CSF component had a mass of 500–3000 Da and was neutralized by trypsin. Flumazenil, a benzodiazepine receptor antagonist, reversed the enhancement of GABAA signaling by hypersomnolent CSF in vitro, and flumazenil normalized vigilance in all seven hypersomnolent patients who underwent the drug challenge [98]. The authors conclude that a naturally occurring substance in CSF augments inhibitory GABA signaling, revealing a new pathophysiology associated with EDS . These results are especially interesting, as GABAAR has never been targeted for the treatment of hypersomnia. It is still unknown if these changes are primary or secondary to the changes in other neurotransmitter systems. It is also critical to test whether the same change is observed in hypocretin-deficient narcolepsy–cataplexy.
Although these new findings are interesting as they are some of the first biomarkers for idiopathic hypersomnia , and these finding may lead to the development of new treatments for somewhat treatment-resistant hypersomnia. However, these markers do not discriminate the types of hypersomnia, and similar changes were observed in various types of hypersomnia.
Symptomatic Narcolepsy and Hypersomnia
Symptoms of narcolepsy can sometimes be seen during the course of a neurological disease process. In such instances, the term “symptomatic narcolepsy” is used, implying that the narcolepsy is a symptom of the underlying process rather than idiopathic. For these cases, the signs and symptoms of narcolepsy must be temporally associated with the underlying neurological process.
In the ICSD-3, narcolepsy with or without cataplexy associated with neurological disorders is classified under “narcolepsy due to medical condition.” The criteria for “narcolepsy due to medical condition” is similar to those for “narcolepsy with cataplexy” and “narcolepsy without cataplexy,” and the diagnostic criteria include (A) the patient must have a complaint of EDS occurring almost daily for at least 3 months. (B) One of the following must be observed: (i) A definite history of cataplexy. (ii) If cataplexy is not present or is very atypical, polysomnographic monitoring performed over the patient’s habitual sleep period followed by an MSLT must demonstrate a mean sleep latency on the MSLT of less than 8 min with two or more SOREMPs. (iii) Hypocretin-1 levels in the CSF are less than 110 pg/mL (or 30 % of normal control values). In addition, (D) a significant underlying medical or neurological disorder must be accountable for the EDS and/or cataplexy, and (E) the hypersomnia is not better explained by another sleep disorder, mental disorder, medication use, or substance use disorder [1]. As mentioned earlier, EDS without cataplexy nor other REM sleep abnormalities is also often associated with these neurological conditions, and is defined as symptomatic cases of EDS (ICSD-3: hypersomnia due to medical condition).
We therefore define “symptomatic narcolepsy” as cases that meet these criteria (if MSLT data were not available, equivalent polygraphic REM sleep abnormalities were also taken into consideration). In addition, an association with a significant underlying neurological disorder that accounts for the EDS and a temporal association (narcolepsy onset should be within 3 years if the causative diseases are “acute” neurologic conditions) are required [100].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


