, A. Imanishi1, Y. Ohmori1, Y. Sagawa1, Y. Takahashi1 , M. Omokawa1, M. Sato1, Y. Hishikawa1, T. Shimizu1, 2 and S. Nishino3
(1)
Department of Neuropsychiatry, Akita University School of Medicine, Akita, Japan
(2)
International Institute for Integrative Sleep Medicine (WPI-IIIS), University of Tsukuba, Tsukuba, Japan
(3)
Sleep and Circadian Neurobiology Laboratory, Center for Narcolepsy, Stanford University School of Medicine, Palo, Alto, CA 94304, USA
Abstract
The symptoms of narcolepsy can occur during the course of other neurological conditions (i.e. symptomatic narcolepsy). We define symptomatic narcolepsy as cases that meet the International Sleep Disorders Narcolepsy Criteria, which are also associated with a significant underlying neurological disorder that accounts for excessive daytime sleepiness (EDS) and temporal associations. By 2005, we have counted 116 symptomatic cases of narcolepsy reported in the literature. As several authors previously reported, inherited disorders (n = 38), tumors (n = 33), and head trauma (n = 19) are the three most frequent causes for symptomatic narcolepsy. A review of these cases (especially those with brain tumors), illustrates a clear picture that the hypothalamus is most often involved. Reduced CSF orexin-A levels were seen in most symptomatic narcolepsy cases of EDS, with various etiologies including brain tumors, head trauma, and immune-mediated neurological conditions, such as neuromyelitis optica (NMO), and EDS in these cases is sometimes reversible with an improvement of the causative neurological disorder and an improvement of orexin status. It is also noted that some symptomatic EDS cases (with Parkinson diseases and the thalamic infarction) were observed,, but were not linked with orexin ligand deficiency. In this chapter, we first provide an overview of cases of symptomatic narcolepsy and EDS and then extend our discussions to the roles of the orexin system in EDS disorders associated with various neurological conditions.
Keywords
NarcolepsyCataplexySleepinessOrexinHypocretinCerebrospinal fluidTumorMultiple sclerosisNeuromyelitis opticAquaporin-41 Introduction
Narcolepsy is a chronic sleep disorder characterized by excessive daytime sleepiness (EDS), cataplexy, hypnagogic hallucinations (HH) and sleep paralysis (SP) (i.e. narcolepsy tetrad) (Nishino et al. 2000; Mignot et al. 2002). A major breakthrough in narcolepsy research was recently made through the identification of orexin deficiency in narcolepsy-cataplexy (Mignot et al. 2002; Nishino et al. 2000, 2001; Dalal et al. 2001; Kanbayashi et al. 2002; Krahn et al. 2002; Bassetti et al. 2003; Ebrahim et al. 2003). Orexins are hypothalamic neuropeptides involved in various fundamental hypothalamic functions including sleep-wake control, energy homeostasis, autonomic and neuroendocrine functions (de Lecea et al. 1998; Sakurai et al. 1998; Willie et al. 2001). Orexin containing neurons are located exclusively in the lateral hypothalamic area (LHA). Since orexin deficiency in narcolepsy is also tightly associated with human leukocyte antigen (HLA) DR2/DQ6 (DQB1*0602) positivity, an acquired cell loss of orexin containing neurons with autoimmune process are suggested in “idiopathic” cases of narcolepsy (Mignot et al. 2002; Kanbayashi et al. 2002). “Idiopathic narcolepsy” is defined as narcolepsy cases unassociated with apparent radiographical or clinical evidence of brain pathology apart from sleep-related abnormalities. Orexin deficiency in the brain can be determined clinically via cerebrospinal fluid (CSF) orexin-A measures with CSF orexin-A levels in healthy subjects above 200 pg/ml regardless of gender, age (from neonatal to 70s), and time of the CSF collections (Nishino et al. 2000, 2001; Kanbayashi et al. 2002). Due to the specificity and sensitivity of low CSF orexin-A levels (less than 110 pg/ml or 30 % of the mean normal levels) narcolepsy-cataplexy is high among various sleep disorders (Mignot et al. 2002; Ripley et al. 2001; Kanbayashi et al. 2003), CSF orexin measures was a diagnostic criteria for narcolepsy-cataplexy in the 2nd edition of international classification of sleep disorders (ICSD-2) (Medicine AAoS 2005). In the 3rd edition of ISCD (ICSD III), narcolepsy was reclassified depending on orexin deficiency status (i.e., Type I and Type II narcolepsy) (Medicine AAoS 2014).
Impaired orexin systems may also be observed in some neurological disorders affecting the LHA (where orexin cell bodies locate) and/or orexin projection pathways. Indeed, Ripley et al. (2001) had measured CSF orexin levels in 235 neurological patients and shown that a subset of subjects with acute or sub-acute neurological disorders (i.e. intracranial tumors, cerebrovascular events, craniocerebral trauma, central nervous system (CNS) infections and Guillain-Barré Syndrome [GBS]) had decreased CSF orexin-A levels, although CSF orexin-A levels in the majority of patients with chronic neurological conditions, such as Alzheimer’s disease and Parkinson disease, were not significantly reduced. Arii et al. (2004) also studied CSF orexin-A levels in 132 pediatric neurological conditions. The results were consistent with Ripley’s study (Ripley et al. 2001), and only a limited number of neurological conditions besides narcolepsy showed reduced CSF orexin-A levels. These included intracranial tumors, craniocerebral trauma, autoimmune and post-infectious diseases [GBS and acute disseminated encephalomyelitis (ADEM)], and some inherited disorders, such as Niemann-Pick disease, type C (NPC) and Prader-Willi syndrome (PWS) (Arii et al. 2004).
These findings are particularly interesting since these neurological conditions are often associated with acutely disturbed consciousness, lethargy, sleepiness, and/or residual sleep disturbances.
In rare cases, symptoms of narcolepsy can be seen during the course of a neurological disease process (i.e. symptomatic narcolepsy). By 2005, we have counted 116 symptomatic cases of narcolepsy reported in the literature and inherited disorders (n = 38), tumors (n = 33), and head trauma (n = 19) are the three most frequent causes for symptomatic narcolepsy (Nishino and Kanbayashi 2005). Involvements of the hypothalamic structures in symptomatic narcoleptic cases have been emphasized repeatedly for many decades (von Economo 1930; Adie 1926), and an impaired orexin system may also be involved in some symptomatic narcolepsy cases. Association with EDS/cataplexy in some inherited neurological diseases (such as NPC, PWS, or myotonic dystrophy) is also known (Kandt et al. 1982; Parkes 1999; Martinez-Rodriguez et al. 2003). An impaired orexin system may thus also be involved in these sleep-related symptoms of these neurological conditions.
In this chapter, we first overview cases of symptomatic narcolepsy reported in literature. Since EDS without other narcolepsy symptoms can also occur with a variety of neurological disorders and are not usually an indication of narcolepsy, we will also extend our discussion on the roles of orexin system in EDS associated with various neurological conditions.
Since data of CSF hypoocretin-1 measures are available for some recent symptomatic narcolepsy and/or EDS cases, we will focus on these cases and discuss the roles of orexin status in these disorders. For this purpose, we categorized the cases as follows: (I) symptomatic narcolepsy-cataplexy associated with focal/generalized CNS invasion, such as cerebral tumors, vascular diseases (Sect. 3.1), and neurodegenerative disorders (Sect. 3.2), (II) hypersomnia associated with (IIa) focal/generalized CNS invasion, such as cerebral tumors, brain infections, vascular diseases, neurodegenerative disorders (AD and PD) and head trauma (Sect. 4.1), and (IIb) with CNS diseases mediated with neuroimmune mechanisms, such as inflammatory and demyelinating diseases (Sect. 4.2). Non-narcoleptic hypersomnia categories include less defined EDS cases, and likely consists of heterogeneous conditions. This is partially due to the fact that applying standardized polygraphic assessments [all night polygraphic recordings followed by multiple sleep latency test (MSLT)] was often difficult in these neurological conditions. However, since prevalence of these hypersomnia cases appeared to be much higher than that of symptomatic narcolepsy, we believe that the discussion on the roles of the orexin system in less well-defined EDS cases also have valuable clinical implications.
2 Definition of Symptomatic Narcolepsy and Its Overview
Symptoms of narcolepsy can sometimes be seen during the course of a neurological disease process. In such instances, the term “symptomatic narcolepsy” is used, implying that the narcolepsy is a symptom of the underlying process rather than being idiopathic. For these cases, the signs and symptoms of narcolepsy must be temporally associated with the underlying neurological process. “Symptomatic narcolepsy” and “secondary narcolepsy” are used more or less indiscriminately, even though they have different meanings. We recommend the use of symptomatic narcolepsy/EDS, since “secondary EDS” has also been used to describe EDS associated with sleep apnea and restless leg syndrome.
In the ICSD-3 (Medicine AAoS 2014), “Narcolepsy due to Medical Condition” is reclassified under “Narcolepsy Type 1 or Type 2 Due to a Medical Condition” depending on the hypocretin deficiency status, and the criteria for “Narcolepsy Type 1 Due to a Medical Condition” is “The condition must fulfill criteria for narcolepsy type 1 (i.e., hypocretin deficient narcolepsy_and be attributable to another medical disorder).” The criteria for “Hypersomnia Due to Medical Condition” has changed to “Hypersomnia Due to a Medical Disorder”. The following are the criteria:
Narcolepsy type 1 (Criteria A and B must be met)
A.
The patient has daily periods of irrepressible need to sleep or daytime lapses into sleep occurring for at least three months.
B.
The presence of one or both of the following:
1.
Cataplexy (as defined under Essential Features) and a mean sleep latency of ≤8 min and two or more sleep onset REM periods (SOREMPs) on an MSLT performed according to standard techniques. A SOREMP (within 15 min of sleep onset) on the preceding nocturnal polysomnogram may replace one of the SOREMPs on the MSLT.
2.
CSF hypocretin-1 concentration, measured by immunoreactivity, is either ≤110 pg/mL or <1/3 of mean values obtained in normal subjects with the same standardized assay.
“Narcolepsy Type 2 Due to a Medical Condition” is “[a] condition [that] fulfills criteria for narcolepsy type 2 and is attributable to another medical disorder.” The following are the criteria:
Narcolepsy type 2 (Criteria A–E must be met)
A.
The patient has daily periods of irrepressible need to sleep or daytime lapses into sleep occurring for at least three months.
B.
A mean sleep latency of ≤8 min and two or more sleep onset REM periods (SOREMPs) are found on a MSLT performed according to standard techniques. A SOREMP (within 15 min of sleep onset) on the preceding nocturnal polysomnogram may replace one of the SOREMPs on the MSLT.
C.
Cataplexy is absent.
D.
Either CSF hypocretin-1 concentration has not been measured or CSF hypocretin-1 concentration measured by immunoreactivity is either >110 pg/mL or >1/3 of mean values obtained in normal subjects with the same standardized assay.
E.
The hypersomnolence and/or MSLT findings are not better explained by other causes such as insufficient sleep, obstructive sleep apnea, delayed sleep phase disorder, or the effect of medication or substances or their withdrawal.
“Hypersomnia Due to a Medical Disorder” fulfills the following criteria.
Hypersomnia Due to a Medical Disorder (Criteria A–D must be met)
A.
The patient has daily periods of irrepressible need to sleep or daytime lapses into sleep occurring for at least three months.
B.
The daytime sleepiness occurs as a consequence of a significant underlying medical or neurological condition.
C.
If an MSLT is performed, the mean sleep latency is ≤8 min, and fewer than two sleep onset REM periods (SOREMPs) are observed.
D.
The symptoms are not better explained by another untreated sleep disorder, a mental disorder, or the effects of medications or drugs.
2.1 Anatomical Substrate for the Symptoms of Narcolepsy
It is important to understand what mechanisms and which brain sites are involved in the occurrence of symptomatic narcolepsy, especially in relation to the orexin system. Although it is not simple to discuss mechanisms uniformly for symptomatic narcolepsy associated with various genetic disorders, analysis of symptomatic narcolepsy with tumor cases showed clearly that the lesions were most often (about 70 % of cases) involved in the hypothalamus and adjacent structures (the pituitary, supraseller or optic chiasm). Impairments in the hypothalamus are noted in most symptomatic cases of narcolepsy which also suggests a possible involvement of impaired orexin neurotransmission.
Lumber CSF orexin-A measurements were carried out in neurological condtions possibly associated with symptomatic cases of narcolepsy/EDS using lumber CSF. The ventricular CSF data of PD in Drouot et al. (2003) were not included. The following is a breakdown of the 346 cases measured by 2014, with incidence of low orexin-A level out of total cases per neurological condition: narcolepsy/EDS associated with tumors (9 out of 14 had low levels of orexin), head trauma (3 out of 7), vascular disorders (1 out of 8), encephalopathies (4 out of 6), neuro degeneration (7 out of 209), immune-mediated demyelinating disorder (11 out of 19), immune-mediated polyneuropathy (1 out of 23), paraneoplastic autoimmune syndrome (2 out of 8), genetic/congenital disorders (7 out of 66) and others (2 out of 4) (Appendix Table). Among the 50 low orexin-A cases, 22 (44 %) cases had hypothalamic lesions and 28 (56 %) cases had no distinct lesions (Fig. 1).
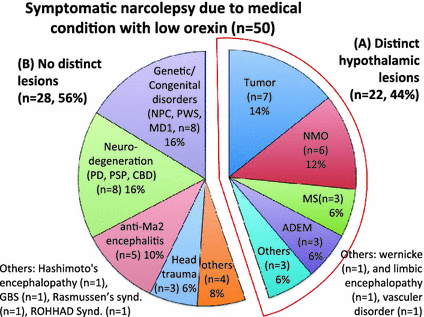
Fig. 1
Category of medical conditions associated with low orexin symptomatic narcolepsy. 50 cases of narcolepsy due to medical conditions with low orexin are included. The percentage of each medical condition was displayed. Tumors (n = 7, 14 %), demyelinating disorders (NMO (n = 6, 12 %), MS (n = 3, 6 %), ADEM, (n = 3, 6 %), genetic/congenital disorders (n = 8, 16 %) and neuro-degeneration (n = 8, 16 %) are the four most frequent causes. Several categories showed distinct hypothalamic lesions (A, n = 22, 44 %), including tumors and demyelinating disorders, while genetic/congenital disorders, neuro-degeneration, paraneoplastic autoimmune syndromes (anti-Ma associated encephalitis) and head trauma did not show distinct lesions (B, n = 28, 56 %)
Recently, we have reported a new possible pathophysiology of symptomatic narcolepsy/EDS in patients with MS and its related disorders (Kanbayashi et al. 2009). These cases often show unique bilateral symmetric hypothalamic lesions associated with significant orexin ligand deficiency. Interestingly, these patients often share clinical characteristics with neuromyelitis optica (NMO) patients, including optic neuritis or spinal cord lesions and the presence of NMO-IgG (anti aquaporin-4 (AQP4) antibodies) (Fig. 2) (Kanbayashi et al. 2009). AQP4 is highly expressed in the hypothalamic periventricular regions (Amiry-Moghaddam et al. 2003; Pittock et al. 2006), thus an immune attack to AQP4 may possibly be responsible for the bilateral and hypothalamic lesions and orexin deficiency in narcolepsy/EDS associated with these diseases. As AQP4 is found in nonneuronal structures such as astrocytes and ependymocytes, impairments of the orexin neurons are likely to be secondary to changes in their surrounding regions (Kanbayashi et al. 2009). None of these cases exhibited cataplexy, but some exhibited REM sleep abnormalities, and thus some of these cases meet the ICSD narcolepsy criteria (Fig. 2). It should also be noted that many earlier narcolepsy-cataplexy cases were associated with MS (5 out of 6 cases reported before 1970). Considering most recent cases were treated with steroids (or other immunosuppressants) at the early stage of the disease and EDS and orexin deficiency was often recovered, chronic impairments of the orexin system may be required for the occurrences of cataplexy (see Nishino and Kanbayashi 2005).
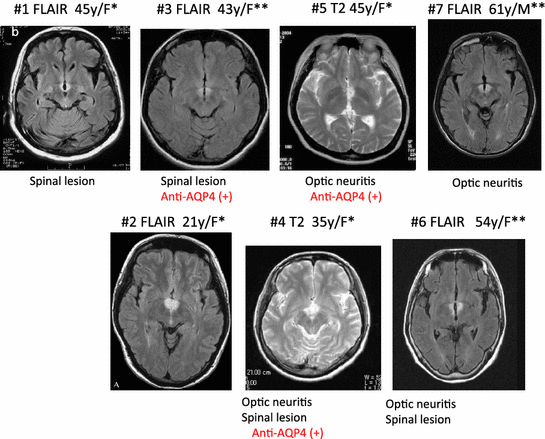
Fig. 2
MRI findings (FLAIR or T2) in multiple sclerosis (MS)/neuromyelitis optica (NMO) patients with orexin deficiency and excessive daytime sleepiness. A typical horizontal slice including the hypothalamic, periventricular area from each case is presented. All cases were female. *, met with ICSD-2 criteria for narcolepsy caused by a medical condition; **, met with ICSD-2 criteria for hypersomnia caused by a medical, condition. All cases were initially diagnosed as MS. Cases 3–7 had optic neuritis and/or spinal cord lesions, and cases 4, 5, and 7 are seropositive for anti-AQP4 antibody and thus were diagnosed as NMO. (Data from Kanbayashi et al. (Kanbayashi et al. 2009)
Although detailed mechanisms of orexin impairments in these NMO subjects need to be further explored, these new finding also confirm the importance of the hypothalamus, where the orexin neurons are located, as the brain structure involved in symptomatic narcolepsy.
3 Orexin Status in Various Neurological Conditions
3.1 Orexin Status in Symptomatic Narcolepsy-Cataplexy Associated with Distinct CNS Lesions
Soon after the discovery of the involvement of orexin impairments in idiopathic narcolepsy, Melberg et al. (2001) reported a reduced CSF hypoocretin-1 level (96 pg/ml) in a previously reported 51-year-old male case with autosomal dominant cerebellar ataxia (ADCA), deafness and narcolepsy (DN). In this Swedish pedigree (ADCA-DN; OMIM, Online Mendelian Inheritance in Man, accession number 604121), four out of five ADCA subjects are affected with narcolepsy-cataplexy (Melberg et al. 1999), and CSF previously collected from one of theses subjects (patients III-2) was available for orexin measures. The patient was negative for HLA-DR2. Since this case is a heredodegenerative disease with an enlargement of the third ventricle, moderate atrophy of the cerebellum and the cerebral hemispheres by MRI were observed, and we listed this case under narcolepsy associated with distinct CNS lesions.
Scammell et al. (2001) subsequently reported a 23-year-old male who developed narcolepsy-cataplexy due to a large hypothalamic stroke after a craniopharyngioma resection. This lesion included 2/3 of the caudal hypothalamus except for the most lateral component on the right and extended into the mediodorsal thalamus bilaterally, the left amygdala, and parts of the basal forebrain and the rostral midbrain. Postoperative course was complicated by panhypopituitarism, staphylococcal meningitis and hydrocephalus. He experienced HH. He became obese with a body mass index (BMI) of 31.7. Sleep latency was 0.5 min according to MSLT, and REM latency was 3.5 min. An overnight polysomnography (PSG) showed 1 min and 1.5 min of SL and REM latency respectively, without significant sleep apnea. HLA was negative for DQB1*0602, and CSF orexin level was 167 pg/ml.
Nokura et al. (2004) reported one case with narcolepsy and cataplexy-like phenomena in a 66-year old female with hypersomnia due to a hypothalamic tumor. She showed EDS and cataplexy-like symptoms, such as abrupt falling without loss of consciousness. An MRI revealed lesions with high signal intensities in the hypothalamus, thalamus and midbrain bilaterally. This case was accompanied with mild anterior hypopituiarism and a SOREMP in a daytime polysomnography. Orexin-A level was 61 pg/ml. Symptoms improved with tumor reduction after radiotherapy and the intravenous administrations of nimustine hydrochloride and interferon beta.
The lesions in these cases had different etiologies: degeneration, infarction and tumor. Although the number of cases is still limited, the hypothalamic lesions were noted in all cases. Moderate reduction of CSF orexin levels (low in two cases and intermediate in one) also confirmed the functional impairment of the hypothalamus. A massive impairment of orexin projections and projection sites are likely involved in the mentioned case with hypothalamic stroke after craniopharyngioma resection, implying that a more severe orexin neurotransmission impairment than the intermediate CSF orexin-A level implies, may exist. Although these results are consistent with the hypothesis of hypothalamic orexinergic involvement in symptomatic cases of narcolepsy, it is not certain if all cases with low orexin levels associated with hypothalamic damage develop narcoleptic symptoms.
3.2 Orexin Status in Symptomatic Narcolepsy-Cataplexy and/or EDS Associated with Inherited Disorders
There are clusters of cases of genetic or congenital disorders associated with primary central hypersomnolence and/or cataplexy, and CSF orexin-A has also been assessed in several patients with Prader-Willi syndrome (PWS), Niemann-Pick type C disease (NPC) and myotonic dystrophy.
3.2.1 Prader-Willi Syndrome (PWS)
EDS is a common symptom in PWS (Vela-Bueno et al. 1984; Helbing-Zwanenburg et al. 1993; Vgontzas et al. 1996). Sleep disordered breathing (SDB) and narcoleptic traits such as SOREMPs and cataplexy have also been reported in these subjects (Manni et al. 2001; Tobias et al. 2002). If SDB exists, primary hypersomnia should only be diagnosed if excessive daytime sleepiness does not improve after adequate treatment of sleep-disordered breathing. Mignot et al. (2002) reported a 16 year old male with the following: EDS, HLA-DQB1*0602 positive, 109 pg/ml orexin-A, obese (BMI = 48.1), documented 15q11-13 deletion, limited number of sleep disordered breathing events (apnea hypoxia index [AHI] = 5.6), no cataplexy, SL = 3.0 min, no SOREMPs by MSLT., Nevsimalova et al. (2004) also measured CSF orexin-A in another three PWS cases. One subject exhibited EDS (AHI = 3.1., age = 10) had low orexin-A levels (130 pg/ml) and DQB1 *0602, but the other two who did not exhibit EDS had intermediate (191 pg/ml) or normal (226 pg/ml) orexin-A with (AHI = 46.8, age = 26) and (AHI = 0, age = 6), respectively. All three subjects were obese and did not exhibit cataplexy. Interestingly, AHI in these PWS subjects were correlated with age and BMI, but not with CSF orexin-A levels and EDS.
Additional reports suggested the possibility that EDS in PWS may also be attributed to the orexin system, not necessarily to sleep disordered breathing caused by obesity. First, Arii et al. (2004) reported a two week-old PWS male with severe hypotonia, poor feeding, documented 15q11-12 deletion and intermediate orexin level (192 pg/ml). Then, Terashima et al. (2012) reported a 11 year-old PWS female with the following: EDS, mildly obesity (BMI = 19.9, %BMI = 108), no sleep disordered breathing events, no cataplexy; SL = 3.0 min, SOREMPs confirmed by MSLT, orexin 60 pg/ml. Dr. Nevsimalova also proposed that PWS cases may be a model for congenital dysfunction/developmental failure of the orexin system (Nevsimalova et al. 2004).
However, no decrease in the number of orexin-containing neurons was observed in post-mortem human adult and infant brains (Fronczek et al. 2005), which suggests a lack of involvement of orexin in the pathogenesis of the disorder. In a larger context, this result underlies the need for larger studies to determine whether decreased CSF orexin-A remains anecdotal in inherited neurological conditions.
3.2.2 Niemann-Pick Type C Disease (NPC)
NPC is an autosomal recessive and congenital neurological disorder characterized by the accumulation of cholesterol and glycosphingolipids in the peripheral tissues and of the glycosphingolipids in the brain. Classic NPC symptoms include hepatosplenomegaly, vertical supranuclear gaze palsy, ataxia, dystonia, and dementia. Subjects with NPC have been reported to frequently display narcolepsy-like symptoms, including cataplexy (Kandt et al. 1982; Autret et al. 1994; Vankova et al. 2003; Vanier 1983; Kanbayashi et al. 2003). This condition is remarkable as cataplexy is often triggered by typical emotions (laughing) and responsive to anticataplectic treatments.
Kanbayashi et al. (2003) measured CSF orexin levels in two NPC cases with and without cataplexy. In the first case (male, age 5), cataplexy and an intermediate orexin level (142 pg/ml) was detected. Cataplexy was triggered by laughter since the age 2. EDS was not claimed by the patient, and SL (16.5 min) was normal without SOREMPs (Philip et al. 1997). No abnormalities in the hypothalamus were detected by MRI scans. He was negative for HLA DR2. In the second case (female, age 3), a normal orexin level (299 pg/ml) was detected. Neurological symptoms such as tremor, ataxia and akathisia were present, cataplexy nor EDS was present.
Vankova et al. (2003) reported five patients with juvenile NPC. Deterioration of intellectual function, the presence of pyramidal, dystonic and cerebellar signs, and splenomegaly were observed in all cases as well as disrupted sleep in nocturnal polysomnography. Total sleep time, sleep efficiency, REM sleep, and delta sleep amounts were decreased when compared to age-matched controls. Cataplexy was reported in one patient. Shortened mean sleep latencies were observed in three patients during the MSLT, but SOREMPs were observed only in the case with cataplexy, and this case met with the criteria of symptomatic cases of narcolepsy. This patient was HLA DQB1*0602 positive, while the other subjects were HLA DQB1*0602 negative. CSF orexin-A levels were reduced in patients (190 pg/ml and 157 pg/ml in the subject with cataplexy) while in the two other patients, the CSF orexin-A were at the lower end of normal (226 pg/ml, 245 pg/ml). The authors speculated that lysozomal storage abnormalities in NPC patients may also have the impact on the hypothalamus including, area orexin-containing cells are located.
Oyama et al. (2006) reported a Japanese patient with NPC caused by a homozygous c.2974 G > T mutation of the NPC1 gene, a well-known NPC1 gene mutation that causes a unique phenotype of NPC, which has been limited to a single Acadian ancestor in Nova Scotia, Canada. The patient characteristically started presenting with cataplexy at the age of 9 years, and the level of orexin-A was moderately low, 174 pg/ml.
Eto et al. (2015) reported NPC case complicated by cataplexy (age 4, male) with orexin level was 106 pg/ml. He had compound heterozygous NPC1 mutations: a novel missense mutation (G9D) in exon 1 and a known missense mutation (R1186H) in exon 23.
Soda et al. (2011) reported a NPC and narcolepsy-cataplexy case (age, 24) with orexin level of 88 pg/ml. Short sleep latency (1 min) and 3 SOREMPs in 4 naps were observed by MSLT. HLA typing was not typical for narcolepsy.
In these five reports, all of the NPC patients with cataplexy have an association with reduced orexin-A levels, while CSF orexin-A levels in the NPC cases without cataplexy are in the lower limit of normal, suggesting a degree of impairments of the orexin system may contribute the occurrence of cataplexy in this inherited diffuse CNS impairment condition.
3.2.3 Myotonic Dystrophy (MYD)
Myotonic dystrophy type 1 (MD1) is a multisystem disorder with myotonia, muscle weakness, cataracts, endocrine dysfunction, and intellectual impairment (Coccagna et al. 1975) (Park and Radtke 1995) (Gibbs et al. 2002). This disorder is caused by a CTG triplet expansion in the 3′ untranslated region of the DMPK gene on 19q13. The expansion resides within ubiquitously expressed genes and when transcribed, accumulates in the nuclei as RNA expansions. This induces the sequestration of muscleblind proteins (Mbnl 1,2,3—RNA binding proteins selective for UG-rich domains) and upregulation of CUG-binding protein/Elav-like family (ex. CELF) resulting in altered splicing of Mbnl-regulated transcripts and causing major aspects of DM (Charizanis et al. 2012; Kanadia et al. 2003, 2006; Hao et al. 2008; Wang et al. 2012; Charizanis et al. 2012). MD1 is frequently associated with EDS and the presence of SOREMPs, which are sleep abnormalities similar to narcolepsy, during the MSLT (Cirignotta et al. 1987; Finnimore et al. 1994; Begin et al. 1997; van der Meche et al. 1994; Guilleminault et al. 1998; Dauvilliers and Laberge 2012) (van Hilten et al. 1993; Gibbs et al. 2002; Hansotia and Frens 1981; Park and Radtke 1995; Yu et al. 2011). The disease is also often associated with SDB, and thus this may also account for appearances of SOREMPs. However, adequate treatment of sleep-disordered breathing does not always eliminate the EDS (van der Meche et al. 1994) (Guilleminault et al. 1998). As many DM1 patients with no sign of sleep apnea or chronic alveolar hypoventilation also exhibit EDS, some authors believe that a central dysfunction is primarily involved in the EDS in DM1 (Dauvilliers and Laberge 2012; van Hilten et al. 1993; Gibbs et al. 2002; Hansotia and Frens 1981).
Martinez-Rodriguez et al. (2003) reported six patients with MYD1 complaining of EDS. The mean sleep latency on MSLTs was abnormal in all patients (<5 min in two, <8 min in four) and two SOREMPs were observed in two subjects, meeting the criteria for symptomatic narcolepsy. It should be noted that these two cases also had SDB. All patients were HLA-DQB1*0602 negative. Orexin-A levels (181 pg/ml) were significantly lower in patients versus controls (340 pg/ml); the one case with two SOREMPs had orexin-A levels in the low range (<110 pg/ml) generally observed in narcolepsy. Three cases had intermediate levels (110–200 pg/ml). The authors suggested that a dysfunction of the hypothalamic orexin system may mediate sleepiness and abnormal MSLT results in patients with MD1.
In one case of late-onset congenital hypoventilation syndrome, a disorder with reported hypothalamic abnormalities (Katz et al. 2000), Martinez-Rodriguez found very low CSF orexin-A levels in an individual with otherwise unexplained sleepiness and cataplexy-like episodes (Martinez-Rodriguez et al. 2003). Excellent response to anti-cataplectic medication was observed in this case.
Iwata et al. (2009) and Yasui et al. (2010) each reported a case of MYD1 with narcolepsy due to medical condition. Both patient had no cataplexy and were HLA-DQB1*0602 negative. In the former case, orexin was markedly decreased to <40 pg/ml. The size of the CTG repeat was markedly increased in the 3′ untranslated region of the DMPK gene at 1800–2400 repeats and PSG revealed severe sleep apnea (AHI = 59/h, BMI = 27.7) and chronic alveolar hypoventilation indicating severe disesase. Since her nocturnal sleeping time was extended to 18 h per day, MSLT revealed normal sleep latencies and no SOREMPs. In the latter case, patient was using Bipap due to nocturnal hypoxia (BMI = 27.3).
However, a larger study failed to confirm these results (Ciafaloni et al. 2008). Orexin-A concentrations did not correlate clinically with disease severity or duration, nor with subjective or objective reports of sleepiness. Because CSF orexin concentrations are often only slightly decreased in some patients, a functional abnormality that causes sleepiness and SOREMPs in myotonic dystrophy type 1 is unlikely to be a common occurrence.
It should also be pointed out that EDS in DM1 is distinctive (from such as that of narcolepsy), and a recent comprehensive sleep evaluations in forty DM1 patients (Yu et al. 2011) demonstrated that unlike in narcolepsy, most patients did not show shortened sleep latency in MSLT (DM1: 14. 2 min (2.8–20 min) versus Control: 14.2 min (8.2–20 min)), although most of them claimed moderate to severe subjective daytime sleepiness (79.5 % vs. 17.1 %, p < 0.002) or fatigue (62.2 % vs. 17.1 %, p < 0.002). The current international criteria for sleep disorders sets the cut off for the MSLT mean sleep latency as less than 8 min, and thus most of these sleepy DM1 patients do not even fit in the diagnostics category of hypersomnias (Medicine AAoS 2005). Occurrence of cataplexy was also never reported in DM1 (Cirignotta et al. 1987; Finnimore et al. 1994; Begin et al. 1997; van der Meche et al. 1994; Guilleminault et al. 1998; Dauvilliers and Laberge 2012; van Hilten et al. 1993; Gibbs et al. 2002; Hansotia and Frens 1981; Park and Radtke 1995; Yu et al. 2011).
Thus, the pathophysiology of EDS in DM1 is truly mysterious.
Recent animal studies using the mouse model of DM demonstrated a selective and robust increase in REM sleep propensity (Charizanis et al. 2012). Mbnl1 KO and Mbnl2 KO mice were recently generated and shown to develop muscle and other DM symptoms, and thus these KO mice are informative animal models of DM (Kanadia et al. 2003; Hao et al. 2008). As Mbnl2 plays a more important role as a splicing regulator during brain development compared to Mbnl1 (Charizanis et al. 2012; Suenaga et al. 2012), the sleep phenotype of Mbnl2 KO mice have been evaluated (Charizanis et al. 2012); Mbnl2 KO mice showed an increase of REM sleep amounts associated with increased EEG theta power. This change was most notable during the dark period when mice are normally awake. Interestingly, a larger portion of these dark period REM sleep episodes in Mbnl2 KOs exhibited a short latency from the proceeding wake episodes, but they did not exhibit cataplexy. A more profound REM sleep rebound after 6 h sleep deprivation was also observed in KOs, compared to wild-type (WT) mice. These sleep changes were REM sleep specific, as no changes in wake and non-REM sleep was seen in these KO mice at the baseline and during sleep rebound, suggesting that Mbnl2 KO mice exhibit selective increases in REM sleep propensity. Based on these results and the fact that selective REM sleep deprivation in human induces a significant increase in REM sleep propensity and sleepiness during daytime (Endo et al. 1998), we hypothesize that abnormal REM sleep propensity may primarily cause EDS in DM1, and altered splicing of Mbnl-regulated transcripts can induce REM sleep abnormalities in DM1.
4 Orexin Status in Hypersomnia in Various Neurological Conditions
4.1 Focal/Generalized CNS Invasion
Symptomatic narcolepsy is relatively rare, but sleepiness without other narcoleptic symptoms can often occur with a variety of neurological disorders; they are more likely to be due to multifocal or global disturbances of the brainstem, diencephalon and cerebral cortex. Recently, several clinical studies also suggested that the disruption of the hypothalamic orexin system in EDS associated with various neurological conditions.
4.1.1 Cerebral Tumors
Cases with EDS seen along with various cerebral tumors have been reported. Six of these cases we reviewed presented low orexin-A levels (Arii et al. 2001; Marcus et al. 2002; Marcus and Mignot 2003; Tachibana et al. 2005; Dauvilliers et al. 2007; Sakuta et al. 2012; Uchida et al. 2014).
(Case 1) Hypersomnia seen after removal of a hypothalamic suprasellar Grade II pilocystic astrocytoma: MRI showed the bilateral, medial and lateral hypothalamic areas and right posterior hypothalamus were damaged. Orexin-A levels was 104 pg/ml and HLA-DR2 negative. Symptoms included diabetes insipidus (DI), hypothyroidism, weight gain, no cataplexy. MSLT: sleep latency: 1.7 min, no SOREMPs (Arii et al. 2001).
(Case 2) EDS in a patient in a vegetative state following astrocytoma resection and CNS hemorrhage: MRI revealed a large suprasellar mass that extended into the sella inferiorly and was displaced posteriorly. Orexin-A was undetectably low and HLA-DR2 and DQB1*0602 was negative. Nocturnal EEG study showed fragmented sleep with 16 short REM cycles. The daytime EEG showed frequent REM periods. EDS improved with 200 mg modafinil and 5 mg methylphenidate (Marcus et al. 2002; Marcus and Mignot 2003).
(Case 3) Hypersomnolence in patient with extensive hypothalamic damage after removal of a craniopharyngioma: CSF orexin-A level (93 pg/ml) was low with negative HLA DQB1*0602 typing.
Short sleep latency and SOREMPs during a MSLT suggested a diagnosis of symptomatic narcolepsy, indicated destruction of orexin-producing neurons in the hypothalamus (Tachibana et al. 2005).
(Case 4) Severe symptomatic narcolepsy in a patient with primary CNS B-cell lymphoma whose symptoms reversed after chemotherapy: MRI revealed an infiltrative hyperintensity in the left basal ganglia, thalamus, cerebral pedunculus, splenium of the corpus callosum, and the right internal temporal lobe. CSF orexin-A level was undetectable and DQB1*0602 typing was negative. Patient had entered a permanent hypersomnia status, related to a coma-like state. After IV and intrathecal chemotherapy, hypersomnia resolved completely and without any abnormal REM sleep manifestation. Eight months later, a 24-h polysomnography was normal without daytime sleep episodes. Brain MRI and FDG PET scans as well as orexin-A level (244 pg/mL), were normal after treatment (Dauvilliers et al. 2007).
(Case 5) A 19-year-old woman suffered from severe EDS accompanied with long sleep episodes both in the daytime and nighttime and frequent episodes of cataplexy shortly after the removal of craniopharyngioma in the intrasellar space (Fig. 3). MSLT showed a typical finding of narcolepsy, and CSF orexin concentration (71 pg/ml) was below the narcolepsy cut-off value. MRI-tractography showed a clear lack of neuronal fiber connections from the hypothalamus to the frontal lobe (Sakuta et al. 2012).
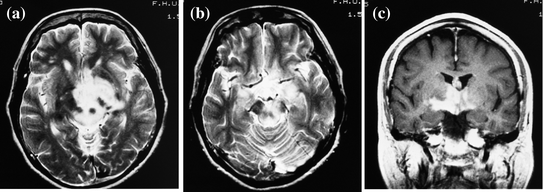
Fig. 3
A narcolepsy-cataplexy case with hypothalamic tumor and low orexin level (61 pg/ml). A 66-year old female case with hypothalamic tumor. a, b Axial T2-weighted image of MRI at admission exhibits high signal intensities in the midbrain, hypothalamus, and thalamus. c Coronal T1-weighted image with gadolinium exhibits enhancement in the same lesion. This case also accompanied with mild anterior hypopituitarism. Her symptoms and MRI findings were improved with reduction of the tumor after 46 Gy radiation and nimustine hydrochloride and interferon beta were administered intravenously (Nokura et al. 2004)
(Case 6) A 13-year-old girl suffered from severe hypersomonolence in the daytime and nighttime and several episodes of cataplexy after the removal of craniopharyngioma. She had also DI and hypothalamic-pituitary dysfunction. Her orexin level was <40 pg/ml (Uchida et al. 2014).
We also reviewed 3 case reports in which orexin-A levels were normal to high.
(Case 7–11) EDS in a total of 5 patients who underwent relatively extensive surgeries involving the hypophysis and hypothalamus, for craniopharyngioma (n = 3), germ cell tumor (1), and thalamic arachnoid cyst (n = 1): The craniopharyngiomas and germ cell tumor were located in the hypothalamus-hypophysis region, and the arachnoid cyst was in the thalamic region. All patients received hormone replacement therapies. Mean orexinn-1 (133 pg/ml) was as same as their control range. The mean sleep latency by MSLT in the five patients was 10.3 min. Two patients were morbidly obese and had obstructive sleep apnea, and although treatment with continuous positive airway pressure resulted in complete resolution of their sleep-disordered breathing, daytime somnolence was unchanged (Snow et al. 2002).
(Case 12) A patient who developed a narcoleptic-like sleep disorder immediately following pinealectomy for a choroid plexus carcinoma of the pineal gland: The patient also underwent chemotherapy and radiation treatment. Immediately after surgery, the patient developed EDS that she attributed to severe insomnia and an irregular sleep/wake rhythm. SP and HH were present but not cataplexy. An increased percentage of REM sleep was seen in nocturnal polysomnography, and three out of four SOREMPs were seen during the MSLT. CSF orexin level (518 pg/ml) was normal, and patient was negative for HLA-DQB1*0602. The author proposed that her symptoms be caused by an unknown mechanism unrelated to orexin depletion (Krahn et al. 2002).
(Case 13) Narcolepsy-cataplexy that developed in acromegaly patient, two weeks after completing radiotherapy for a pituitary adenoma: Orexin-A was normal (275 pg/ml) and HLA was not typical for narcolepsy. Both HH and SP were present. Sleep latency by MSLT was 6.4 min and REM latency was 9 min (3 SOREMPs/5 naps). He was obese (BMI: 35) and his AHI was 17/h. The authors have speculated that the radiotherapy of the tumor was associated with damage to a locus rich in orexin receptors (Dempsey et al. 2003).
Overall, we reviewed 7 symptomatic cases with EDS with low orexin-A levels (one case in Sect. 3.1) and 7 cases in 3 case reports with normal to high orexin-A. All the cases with low CSF orexin-A levels were either HLA-DR2 or HLA-DR2 and DQB1*0602 negative, thus EDS in these cases are likely secondary to the orexin deficiency caused by the tumors/tumor removal. Other mechanism likely cause EDS in the 7 cases with normal or high orexin-A levels, although impairment in orexin projections, terminals or postsynaptic receptors may also be caused by the tumors.
4.1.2 Infarctions
EDS has been reported in cerebra infraction cases. Bassetti et al. reported two cases with EDS and cerebral infarction. In a thalamic infarction case, mean sleep latency was 9 min and orexin level was 265 pg/ml. In a ponto-medullary infarction case, sleep latency was 1 min and orexin level was 316 pg/ml (Bassetti et al. 2003).
Two hypersomnia cases with bilateral paramedian thalamic infarctions were also independanlty reported (Nokura et al. 2004; Tohyama et al. 2004). The paramedian thalamus believed to play an important role in the regulation of sleep, and disturbances of sleep regulation are known to occur in paramedian thalamic stroke (Guilleminault et al. 1993; Bassetti et al. 1996). The first case suffered from bilateral paramedian thalamic infarctions and had EDS with SOREMPs (two times in four naps). His orexin-A level was 312 pg/ml (Nokura et al. 2004), the symptoms met with the criteria for Narcolepsy Type 2 Due to a Medical Condition. The second case suffered from bilateral paramedian thalamic infarctions and hypersomnia. His orexin level was 274 pg/ml (Tohyama et al. 2004) (Fig. 4). Since the lesions of infarctions did not include the orexin cell bodies, their orexin levels seemed to be normal. However, orexin projection could still be impaired. Guilluminault et al. has pointed out that patients with bilateral paramedian thalamic lesions do not present a typical hypersomnia but a de-arousal or subwakefullness with an inability to develop sleep outside the normal circadian boundary (pseudo-hypersomnia) (Guilleminault et al. 1993). Indeed these patients showed reduced latency to stage 1 during MSLT, but did not develop other normal non REM sleep and REM sleep status during the daytime. It may also be possible that orexin deficiency is not involved in so-called pseudo-hypersomnia associated with bilateral paramedian thalamic lesions, and other pathophysiology needs to be considered for these unique sleep symptoms.
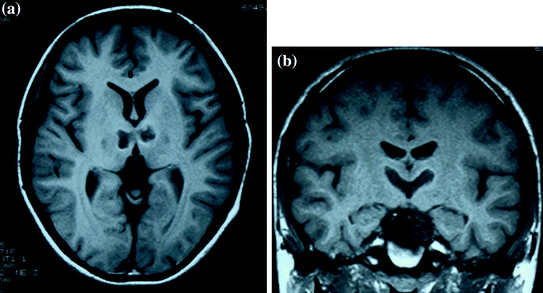
Fig. 4
a, b A 15 year old case with paramedian thalamic infarctions and normal orexin level (274 pg/ml) (Tohyama et al. 2004). Dauvilliers et al. (2007) reported a case with paramedian thalamic infractions and normal orexin level (274 pg/ml). A 15-year old male with EDS due to bilateral paramedian thalamic infarctions. Patients with bilateral paramedian thalamic lesions are known to often exhibit atypical hypersomnia (i.e. de-arousal or subwakefullness) (Sakuta et al. 2012). The lateral hypothalamus (where orexin cell bodies locate) was not affected, and CSF orexin level was in the normal range. It is not known whether the other orexin systems (projections or receptive sites) are still involved in EDS with paramedian thalamic infractions
4.1.3 Encephalopathies
Wernicke’s Encephalopathy
A five-year-old female with Wernicke’s Encephalopathy (Fig. 5) was reported to have gradually developed sleepiness and an abnormal sleep/wake schedule (Kashiwagi et al. 2004). She slept 15–20 h per day and fell asleep frequently even while eating. She developed ocular and neurological symptoms (such as involuntary movements, hemiparesis, depression of speech and global confusional state). An MRI revealed lesions in the bilateral hypothalamus in addition to the dorso-medial nucleus of thalamus and mammillary bodies and periaqueductal gray and floor of 4th ventricle. Vitamin B1 levels were low (38.7 ng/ml, normal range: 52–176 ng/ml) and the level of orexin of CSF was decreased (<40 pg/ml). Her sleepiness and MRI findings gradually improved with thiamine therapy. Six months after the onset of sleepiness, both MRI lesion and CSF orexin level (158 pg/ml) recovered to some degree. It is not clear whether Wernicke’s encephalopathy affect the orexin system directly or indirectly. It is also not fully studied whether the change in the orexin neurotransmission is solely responsible for the occurrence of the EDS. The dysfunction of orexin neuron due to hypothalamic lesion would be caused by damages for AQP4 water channel (Chan et al. 2004).
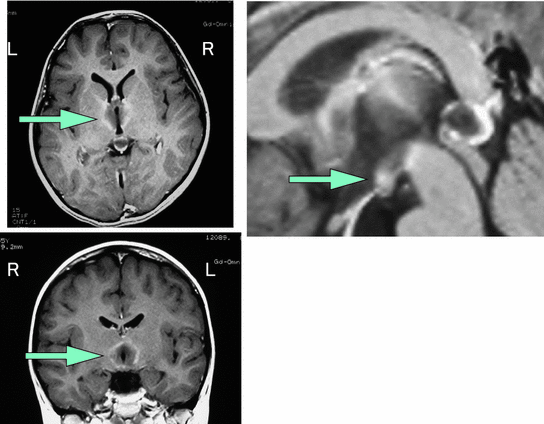
Fig. 5
Hypersomnia due to Wernicke encephalopathy. A 5 year old case with Wernicke’s encephalopathy. Her sleep time was 15–20 h per day and she fell asleep frequently even while eating. Gd-enhanced MRI revealed lesions in the bilateral hypothalamus in addition to dorso-medial nucleus of thalamus and mammillary bodies and periaqueductal gray and floor of IVth ventricle. The level of vitamin B1 was low. The level of orexin was decreased (<40 pg/ml). Her sleepiness and MRI findings gradually improved with replacement of vitamin B1 (Kashiwagi et al. 2004)
Limbic Encephalopathy
Chronic progressive hypersomnia was seen in a patient with non-paraneoplastic immune-mediated limbic encephalitis. Orexin-A concentration was low (87 pg/ml). An MRI of the brain showed bilateral signal abnormalities in the medial temporal lobes and the hypothalamus, but systemic examinations for malignant tumors were negative. Acyclovir treatment failed to amend his condition. Subsequent steroid treatment improved his hypersomnia and reduced the extent of abnormal signals on MRI. The CSF orexin concentration increased to 148 pg/ml in 23 days after (Yamato et al. 2004).
Rasmussen’s Syndrome
Lagrange et al. reported a case of narcolepsy and Rasmussen’s syndrome in a previously healthy 40 year-old man. Severe EDS, cataplexy, HH, and SP developed over the course of a few months. Brain MRI was normal and polysomnography with MSLT confirmed a diagnosis of narcolepsy (SL: 1.6 min, three SOREMPs in four naps). His HLA haplotype is DQB1*0602, and CSF analysis showed no detectable orexin. Approximately 18 months later, he developed complex partial seizures. Further MRI showed a progressively enlarging lesion involving the left frontotemporal and insular areas. Pathology from a partial resection samples was consistent with Rasmussen’s syndrome. Evaluation for tumor, infectious, and paraneoplastic etiologies was negative. There was no further progression of the residual lesion on serial MRI (Lagrange et al. 2003).
Although the pathophysiological bases of narcolepsy and Rasmussen’s syndrome are unknown, the author speculated the possibility of a common underlying disease processes related to autoimmune mechanism. However, whether or not this case highlighted a temporal relationship between orexin deficiency and the onset of the disease is not known. It nay also be possible for Rasmussen’s syndrome to be the comorbidity with idiopathic narcolepsy, since the subject is HLA positive, and late onset cases of idiopathic narcolepsy are also reported.
Brain Stem Encephalitis
Mathis et al. described a case of a previously healthy young man who concurrently developed a narcoleptic syndrome and a fullblown REM sleep related behavior disorder (RBD) after an acute brain stem encephalitis with an isolated inflammatory lesion in the dorsomedial pontine tegmentum. Presenting with hypersomnia, sleep paralysis, hypnagogic hallucinations and SOREMPs, the patient fulfilled the criteria of narcolepsy, although cataplexy was mild and rare. CSF orexin was normal (266 pg/ml) and HLA haplotypes were not typically associated with narcolepsy and RBD (DQB1*0602, DQB1*05) (Mathis et al. 2007).
Hashimoto’s Encephalopathies
Castillo et al. (2006) reported a 65 years old male patient with steroid-responsive encephalopathy associated with autoimmune thyroiditis (SREAT) and hypersomnolence and then coma. He had undetectable levels of orexin-A in the CSF during symptomatic period.
4.1.4 Neurodegenerative Disorders
Parkinson’s Disease (PD)
Thirty percent of patients with Parkinson disease (PD) have been reported to have EDS. Sleep problems are often related to the disease itself (e.g., difficulties in maintaining sleep because of motor disabilities), but they can also occur secondary to pharmacological treatments, especially with dopamine D2/3 agonists. Ripley et al. initially reported that CSF orexin-A in 7 PD subjects were in the normal range, but sleep abnormalities of these subjects were not assessed (Ripley et al. 2001). In a separate study, CSF orexin levels were also normal in all three PD patients with EDS (Overeem et al. 2002).
However, subsequent studies reported that patients with late-stage PD had low ventricular CSF orexin-A levels (n = 16: < 50-97 pg/ml, N = 3: 138-169 pg/ml) (Drouot et al. 2003). Orexin-A levels decreased with increasing disease severity. The author described that CSF orexin-A levels may reflect the size of the orexin neuron pool, and a decrease in orexin-A level may indicate degeneration of orexin neurons in PD. The sleepiness of the patients was assessed by Epworth sleepiness scale (ESS). The mean ESS of these PD patients (11 ± 1) was significantly higher than that of controls (4 ± 1), but orexin-A level was not correlated with ESS among PD subjects.
Two recent studies reported significant (50 %) orexin cell loss in post-mortem hypothalami of patients with Parkinson’s disease, and the presence of Lewy bodies in some orexin-producing cells (Fronczek et al. 2007; Thannickal et al. 2007). In Parkinson’s disease, orexin cell loss is 23–62 % and correlates with disease severity (Thannickal et al. 2007), as measured on the Hoehn and Yahr scale (Hoehn and Yahr 1967). However, orexin cell loss was not specific, and nearby neurons containing melanin-concentrating hormone were similarly lost (12–74 %) in proportion to disease severity (Thannickal et al. 2007). Furthermore, CSF orexin-A levels were normal in Parkinson’s disease (Ripley et al. 2001; Dauvilliers et al. 2003; Overeem et al. 2002; Yasui et al. 2006; Asai et al. 2009; Compta et al. 2009), even if associated with severe sleepiness (Overeem et al. 2002; Baumann et al. 2005), in most studies, excluding a few (Drouot et al. 2003, 2011; Maeda et al. 2006; Mizuno et al. 2011; Wakai and Kanbayashi 2011; ) (Teraoka et al. 2013). Interestingly however, significant reductions in the number of orexin cells in the hypothalamus (Fronczek et al. 2007; Thannickal et al. 2007) and decrease in orexin-A concentrations in the ventricles (Drouot et al. 2003; Fronczek et al. 2007) are evident, and thus moderate orexin deficiency that are not detectable by lumbar CSF orexin-A measures likely exist in a subset of PD subjects. Specificity of these finding and functional correlations, especially with EDS are still unknown.
Dementia with Lewy Bodies (DLB)
Dementia with Lewy bodies (DLB) is the second major type of senile, degenerative dementia, after Alzheimer’s disease (AD). DLB shares many features with PD. EDS, hallucinations and REM sleep behavior disorder are symptoms reported in both DLB and narcolepsy. However, Baumann et al. (2004) reported that patients with DLB had normal orexin-A levels. No histological studies focusing on orexin neurons in DLB was available.
CSF orexin-A concentrations have also been assessed in multiple system atrophy, DLB and corticobasal degeneration (Ripley et al. 2001; Dauvilliers et al. 2003; Friedman et al. 2007; Baumann et al. 2004; Yasui et al. 2006; Martinez-Rodriguez et al. 2007; Abdo et al. 2008). In almost all cases, CSF orexin-A concentrations were normal.
Progressive Supranuclear Palsy (PSP)
EDS was reported in probable progressive supranuclear palsy (PSP) in a 74 year-old female. The EDS mimicked narcolepsy without cataplexy (MSLT showed short latencies of less than 2 min without SOREMPs), HLA was positive for DR2/DQB1 and CSF orexin-A concentration was undetectable (Hattori et al. 2003). It is not clear if the co-occurrence of these disorders is due to a common process or comorbidity. The author speculated that the existence of neuropathological changes, such as neurofibrillary tangles in hypothalamus of the patient with PSP might cause decreased orexin neurotransmission.
Narcolepsy with cataplexy has also been reported in probable PSP, in a 74 year-old male (Sugiura et al. 2007). Patient medical history revealed cataplexy and EDS symptoms fluctuating since age 20, but by age 69, cataplexy and EDS had returned. PSP symptoms such as dysarthria, difficulty in writing, gait disturbance, appeared from age 70. At the time of detailed examinations, ESS was 14 and patient showed the sleep paralysis even during eating and the cataplexy induced by laughter. Other symptoms included the following: vertical gaze limitation, Meyerson sign, small voice, a masked face, wide gait, easily falling backwards, mild muscular rigidity in neck and wrists, bradykinesia in the extremities, no resting tremor, these symptoms were agreeable to diagnose as PSP. In the MRI, the third ventricular enlargement, midbrain tegmentum atrophy and mild frontal lobe atrophy were detected. CSF orexin-A was less than 40 pg/ml and HLA DR2 and DQB1 were positive. In PSG, total sleep time was short with 274 min, 43.1 % wake after sleep onset were present. The AHI was 14/h. Mean sleep latency was shortened to less than 2.9 min and SOREMPs were present in all four naps. L-Dopa and Amantadine were slightly effective for gait, but never effective for other motor symptoms. Methylphenidate (20 mg/day) was effective for daytime sleepiness and Clomipramine was effective for cataplexy.
Yasui et al. also reported that orexin levels were significantly lower in the PSP group compared to PD (p < 0.001) and that orexin levels were inversely correlated with duration of morbidity in PSP but not in the other conditions studied (Yasui et al. 2006). They speculated that loss of orexin neurons or impaired orexin neurotransmission might exist as a part of the neurodegeneration associated with advanced PSP with long duration of morbidity. Considering the aforementioned two case reports by Hattori et al. (2003) and Sugiura et al. (2007), PSP may be a susceptibility factor for EDS and/or symptomatic narcolepsy associated with orexin deficiency. However, more cases are needed to address this question.
Alzheimer’s Disease (AD)
CSF orexin-A levels in 24 patients with Alzheimer disease (AD) were reported normal (Ripley et al. 2001). AD was known with established sleep abnormalities (Bliwise et al. 2002). In AD subjects, dysfunction of other neurochemical systems, for example cholinergic systems, may be more directly involved in sleep abnormalities.
Subsequently, several studies have explored orexin abnormalities in Alzheimer’s disease. Studies in older rats have suggested a very slight orexin cell loss and significantly decreased CSF orexin-A concentrations (Desarnaud et al. 2004). By contrast, lumber CSF orexin-A concentrations have been shown to be normal in all studied patients with Alzheimer’s disease (Dauvilliers et al. 2003; Ebrahim et al. 2003; Friedman et al. 2007), although wake fragmentation was correlated with lower CSF orexin-A concentrations in one study (Friedman et al. 2007). No histological studies focusing on orexin neurons in Alzheimer’s disease were available.
Hungtington’s Disease
In Huntington’s disease, disrupted orexin transmission was first suggested through the study of R6/2 mice, a murine model of Huntington’s disease with accelerated disease progression. Low CSF orexin-A concentrations and decreased orexin cell counts were reported in these mice (Petersen et al. 2005). Huntington’s disease is an autosomal dominant disorder with impaired motor coordination, caused by a CAG triplet repeat extension in the Huntington’s disease gene (HTT). Huntington’s disease is not associated with hypersomnia, cataplexy, or SOREMPs. Widespread cell loss occurs in Huntington’s disease, including in the hypothalamus (Petersen and Bjorkqvist 2006). A slight (27 %) loss of orexin neurons was also reported in post-mortem human brains (Petersen et al. 2005). More recent studies have shown that the cell loss is not associated with low CSF orexin-A concentrations (Baumann et al. 2006; Bjorkqvist et al. 2006; Gaus et al. 2005; Meier et al. 2005). Functional roles of orexin cell loss in Huntington’s disease is not known, but may not strong. Indeed, studies in rats have shown that decreased CSF orexin occurs only when more than 50 % of cells are lost or affected (Gerashchenko et al. 2003; Zhang et al. 2007).
4.1.5 Head Trauma
The association of narcolepsy/EDS with head injury is controversial. Most people with hypersomnolence after closed head injury do not have narcolepsy (Guilleminault et al. 1983), but some patients with narcolepsy report that their symptoms began after a head injury (Lankford et al. 1994; Good et al. 1989; Maccario et al. 1987; Francisco and Ivanhoe 1996; Maeda et al. 1995; Bruck and Broughton 2004). Lankford et al. (1994) reported 9 detailed cases with narcolepsy (5 HLA positive, 2 HLA negative and 2 undetermined), but orexin-A levels were not measured. Later, low to undetectable CSF orexin-A concentrations have been found in many patients with acute brain trauma or post-CNS haemorrhage (Ripley et al. 2001; Baumann et al. 2005; Dohi et al. 2008). Because adding blood to CSF in vitro does not alter CSF orexin-A concentrations, the possibility of a functional connection has been raised.
Dauvilliers et al. (2003) reported that a patient severely affected with post-traumatic hypersomnia with brain lesions (determined by MRI) had an intermediate CSF orexin-A level (176 pg/ml, HLA negative), while another severely affected patient had a normal level (503 pg/ml, HLA positive). These two patients had no cataplexy but had shortened sleep latencies (4.5 min, 3.0 min, respectively) without SOREMPs by MSLT.
Arii et al. (2004) reported a 15-year-old male affected with post-traumatic hypersomnia with an intermediate orexin-A level. His Glasgow scale at 48 h after injury was 12 (E2V4M6). An MRI showed severe cerebral contusion of the bilateral basalis of the fronto-temporal lobe and medial part of right occipital lobe with CSF leakage. One year after injury, he needed more than nine hours nocturnal sleep and one or two 1–3 h naps daily. The orexin-A level was 151 pg/ml. MRI showed atrophies in the basalis of temporal lobe and medial part of right occipital lobe. The hypothalamus showed moderate atrophy with dilatation of third ventricle but no localized lesion.
Baumann et al. (2005) reported abnormally low CSF orexin-A concentrations immediately after traumatic brain injury in approximately 95 % of patients with severe-to-moderate brain injury. However, orexin-A concentrations improved to normal in most patients 6 months after traumatic brain injury, suggesting a functional alteration rather than neuronal loss (Baumann et al. 2007). Further studies are assessing the prevalence of residual hypersomnia and narcolepsy in correlation with CSF orexin-A concentration and areas of focal damage. A temporary decrease in CSF orexin-A could indicate a decrease in orexin tone (e.g., if CSF flow dynamics or dilution occurs) and/or contribute to changes in consciousness in patients with traumatic brain injury.
Baumann reported two male patients in whom MSLT revealed >2SOREMP’s and abnormally short mean sleep latencies (6.3 and 2.9 min, respectively) (Baumann et al. 2007). ESS scores were 13 and 9, respectively. In both patients, Ullanlinna and Swiss Narcolepsy Scales were normal. Neither patient had cataplexy-like episodes, hypnagogic hallucinations, or sleep paralysis. CSF orexin-A levels in the acute phase were 63 and 83 pg/ml. Six months after TBI, levels were normal (468 pg/ml) and low (289 pg/ml), respectively. HLA typing was negative for both patients. In the younger patient, TBI was mild, but severe in the 26-year-old patient. Brain CT scans did not reveal hypothalamic lesions. These patients were asymptomatic before TBI. Based on the MSLT findings and according to the international classification of sleep disorders, these two patients can be diagnosed as narcolepsy without cataplexy [ICSD2] (Medicine AAoS 2005).
One male patient (22 years old) reported hypnagogic hallucinations and cataplexy-like episodes (subjective weakness in both knees with laughter), which did not fulfill the criteria of cataplexy (Anic-Labat et al. 1999). ESS was 11, Ullanlinna Narcolepsy Scale 15, Swiss Narcolepsy Scale normal, mean sleep latency 5.6 min and there were no SOREMP’s. This patient with a narcolepsy- cataplexy-like phenotype reported that he had not observed these symptoms prior to TBI. CSF orexin-A was low 6 months after TBI (225 pg/ml). There were two other patients with a low CSF orexin-A level 6 months after TBI (besides one patient with narcolepsy, and one patient with a narcolepsy-like phenotype, see earlier). In a 58-year-old patient (211 pg/ml), PSG revealed a moderate sleep apnea syndrome (apnea hypopnea index: 25/h), and a short mean sleep latency on MSLT (2.5 min). In a 19-year-old patient (234 pg/ml), all findings were normal.
EDS appearing during the first year following a head injury may be considered as post-traumatic (Billiard 2003). This typically presents itself as extended night sleep and episodes of daytime sleep. Sleepiness is usually associated with other characteristics such as headaches, difficulties in concentration or memory disorder. Radioimaging studies may reveal several possibilities: lesions affecting the hypothalamic region or brainstem, midbrain or pontine tegmentum, or more often than not, the absence of any significant lesions. Sleepiness should be objectively evaluated by a MSLT but is often is not in clinical situation. Cases with hypersomnia after head or brain trauma associated with sleep apnea syndrome were also reported (Guilleminault et al. 1983).
Although two out of three patients with post-traumatic EDS had decreased CSF orexin-A levels moderately, it is not known whether all post-traumatic subjects with declined CSF orexin-A levels exhibit EDS. Similarly, it has not been studied whether more pronounced degree of orexin-A impairments is evident for the post-traumatic symptomatic narcolepsy.
4.2 CNS Diseases Mediated with Neuroimmune Mechanisms
In this section, we will specifically discuss neuro-immunological disorders that meet the ICSD3 criteria of “Narcolepsy Due to Medical Conditions”. There are three reasons for discussing this topic; (1) The etiology of idiopathic (orexin deficient) narcolepsy is not yet known, but an involvement of neuroimmune-interaction is suggested, (2) functional significance of CSF orexin levels in symptomatic narcolepsy and symptomatic EDS has not been evaluated systematically, and (3) our recent study suggests an existence of a new clinical syndrome, symptomatic EDS associated with neuromyelitis optica (NMO) and with anti-aquaporin 4 (AQP4) antibody, with low CSF orexin-A levels. Symptomatic narcolepsy cases with NMO and/or MS cases with anti-aquaporin 4 (AQP4) antibody cases are extremely interesting both in the clinical practice and research. Some of these cases were previously categorized as a Multiple Sclerosis (MS) subtype, and our findings may explains why some of MS cases show EDS and selective lesions in the paramedian hypothalamus and periventricular area.
We will include clinical data of ‘Narcolepsy Due to medical Condition’ from the following three subcategories, (1) Acute disseminated encephalomyelitis (ADEM) (2) Multiple Sclerosis (MS), (3) Neuromyelitis optica (NMO) and Anti-aquaporin 4 (AQP4) antibody.
4.2.1 Acute Disseminated Encephalomyelitis (ADEM)
Symptomatic narcolepsy was recently reported in four ADEM cases (Kubota et al. 2002; Gledhill et al. 2004; Yoshikawa et al. 2004; Mizuno et al. 2011; Yano et al. 2004). All these cases associated with EDS had hypothalamic lesions and low CSF orexin-A levels, suggesting an involvement of the hypothalamic orexin system in these conditions.
Improvement of sleepiness and increase in orexin-A levels after treatment has also been reported in ADEM patients. A 38 year-old female had hypersomnia, but no REM related symptoms such as cataplexy, hypnagogic hallucinations, or sleep paralysis. An MRI revealed lesions in the hypothalamus, walls of 3rd ventricle, corona radiata, floor of the aqueduct, and raphe nuclei. She was positive for DR2/DQB1*0602 and orexin-A levels were 87 pg/ml. After treatement with high-dose steroids, MRI showed smaller and fewer lesions. Six months later, her subjective sleepiness was partially improved and orexin-A level was 148 pg/ml. One year after her initial examination, her sleepiness persisted and the results of MSLT were almost unchanged (Gledhill et al. 2004).
In a 7-year-old girl with ADEM, visual symptoms, and hypersomnia, MRI revealed bilateral lesions in the white matter, basal ganglia, and hypothalamus. CSF orexin-A level was intermediate (146 pg/ml) at admission, and with steroid plus treatment, the orexin level gradually recovered to the normal range (263 pg/ml) within 47 days, and excessive sleepiness was reduced. Decreased hypothalamic orexin neurotransmission may be involved in this symptomatic case of hypersomnia associated with a clinical course of ADEM, and interestingly, double vision was also noted in this case during the course of the disease (Yoshikawa et al. 2004).
4.2.2 Demyelinating Diseases: Multiple Sclerosis (MS) and Neuromyelitis Optica (NMO)
Symptomatic narcolepsy in patients with MS have been reported for several decades. Because both MS and narcolepsy are associated with the HLA-DR2 positivity, an autoimmune target on the same brain structures has been proposed to be a common cause for both diseases, (Poirier et al. 1987). However, the discovery of the selective loss of hypothalamic orexin neurons in narcolepsy indicates that narcolepsy coincidently occurs in patients with MS when MS plaques appear in the hypothalamic area and secondarily damage the orexin neurons. Supporting this interpretation, the orexin system is not impaired in patients with MS who do not exhibit narcolepsy (Ripley et al. 2001). Nevertheless, a subset of patients with MS predominantly shows EDS and REM sleep abnormalities, and it is likely that specific immune-mediated mechanisms may be involved in these cases.
Kanbayashi et al. recently reported 7 cases of EDS occurring in patients initially diagnosed with MS with symmetric hypothalamic inflammatory lesions and orexin ligand deficiency that contrasts with the typical MRI image of MS (Fig. 2). CSF orexin measures revealed that marked (<110 pg/mL, n = 3) or moderate (110-200 pg/mL, n = 4) orexin deficiency was observed in all 7 cases (Kanbayashi et al. 2009). Four of these cases met with ICSD-2 criteria (Medicine AAoS 2005) for narcolepsy caused by a medical condition, and 3 cases met criteria for hypersomnia caused by a medical condition. HLA was negative for DQB1*0602 in the 2 cases evaluated for it. Orexin evaluation was repeated in 6 cases, and CSF orexin-A levels became normal or significantly increased, along with marked improvements of EDS and hypothalamic lesions in all cases (Kanbayashi et al. 2009). Because four cases had clinical characteristics of neuromyelitis optica (NMO) (either optic neuritis and spinal cord lesions, or both, were present) anti-AQP4 antibody was evaluated, and 3 cases came back positive; these were diagnosed as NMO-related disorder.
AQP4, a member of the AQP superfamily, is an integral membrane protein that forms pores in the membrane of biologic cells (Amiry-Moghaddam et al. 2003). Aquaporins selectively conduct water molecules in and out of the cell while preventing the passage of ions and other solutes, and are known as water channels. AQP4 is expressed throughout the central nervous system, especially in periaqueductal and periventricular regions (Amiry-Moghaddam et al. 2003; Pittock et al. 2006), and is found in nonneuronal structures such as astrocytes and ependymocytes, but is absent from neurons. NMO-IgG, which can be detected in the serum of patients with NMO, has been shown to selectively bind to AQP4 (Lennon et al. 2005). Because AQP4 is enriched in the periventricular regions of the hypothalamus, where orexin-containing neurons are primarily located, symmetric hypothalamic lesions associated with reduced CSF orexin-A levels in our 3 NMO cases with anti-AQP4 antibody might be caused by the immune-attack to the AQP4 that secondarily affect the orexin neurons. However, as described earlier, Kanbayashi et al. also had 4 MS cases with EDS and orexin deficiency that tested negative for anti-AQP4 antibody,, which leaves a possibility that other antibody-mediated mechanisms are additionally responsible for the bilateral symmetric hypothalamic damage causing EDS in the MS/NMO subjects. There is also a possibility that these 4 MS cases could be NMO, because anti-AQP4 antibody was tested only once for each subject during the course of the disease and the assay was not standardized among the institutes (Kanbayashi et al. 2009).
It is thus essential to further determine the immunologic mechanisms that cause the bilateral hypothalamic lesions with orexin deficiency and EDS, and their association with NMO and AQP4. This effort may lead to establishment of a new clinical entity, and the knowledge is essential to prevent and treat EDS associated with MS and its related disorders. None of these cases had cataplexy, contrary to the 9 out of 10 symptomatic narcoleptic MS cases reported in the past (Nishino and Kanbayashi 2005). Early therapeutic intervention with steroids and other immunosuppressants may thus prevent irreversible damage of orexin neurons and chronic sleep-related symptoms.
4.2.3 Guillain-Barre’s Syndrome (GBS)
Guillain-Barré syndrome (GBS) is an acute autoimmune polyradiculoneuritis with sensory and motor impairment. Since GBS may also cause autonomic dysfunction, aspiration pneumonia, and respiratory failure, some patients undergo intensive care including invasive ventilation. Although GBS is generally restricted to the peripheral nervous system, clinically and pathologically, central dysfunctions have also been documented (Cochen et al. 2005). These include hyponatremia caused by abnormal antidiuretic hormone secretion (Hochman et al. 1982), rapid eye movement sleep (REM sleep) motor behavior disorders (Schenck et al. 1986), EDS (Guilleminault and Mondini 1986), and abnormally low CSF orexin-A levels (Ripley et al. 2001; Kanbayashi et al. 2002; Nishino et al. 2003). A subset of Miller-Fisher syndrome subjects, but not chronic inflammatory demyelinating polyneuropathy (CIDP) subjects, also has significantly low CSF orexin-A (Nishino et al. 2003).
Undetectably low CSF orexin-A levels were found in seven cases of GBS in the Japanese population (Ripley et al. 2001; Kanbayashi et al. 2002; Nishino et al. 2003). Reduced CSF orexin-A levels in GBS are not likely due to secondary effects of the treatment or associated health conditions, since two GBS patients showed undetectable levels at the time of admission to the hospital (before treatment), but only exhibited general fatigue and/or lower limb weakness, with no increase in CSF protein 1evels (Nishino et al. 2003).
Related posts:
 and Metabolism
Regulates Glucose Homeodynamics with Daily Rhythm
and Output Systems of Orexin Neurons
(Orexin) Cell Transplantation as a New Therapeutic Approach in Narcolepsy
Pathology in Human Narcolepsy with and Without Cataplexy
of Neuronal Circuitry Involved in the Regulation of Sleep/Wakefulness Using Optogenetics
and Metabolism
Regulates Glucose Homeodynamics with Daily Rhythm
and Output Systems of Orexin Neurons
(Orexin) Cell Transplantation as a New Therapeutic Approach in Narcolepsy
Pathology in Human Narcolepsy with and Without Cataplexy
of Neuronal Circuitry Involved in the Regulation of Sleep/Wakefulness Using Optogenetics
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


