Fig. 2.1
Schematic diagram of NSC/NPC behavior in the RMS. a NSC/NPC behavior in the RMS of intact brain. Note that NSCs/NPCs in the RMS are divided into two sub-types: bridge NSCs/NPCs (green) and migrating NSCs/NPCs (pink). The bridge NSCs/NPCs are not traveling cells, and their biological function appears to generate a “bridge” between the olfactory bulb (OB) and subventricular zone (SVZ) for supporting NSC/NPC migration. The migrating NSCs/NPCs are the traveling cells in the RMS, and their traveling directions are multiple (purple arrows). b – f NSC/NPC behavior in the RMS of the brain suffering from cortical ischemic injury during 3–18 h after the induction of cortical brain ischemia. Note that there are no migrating NSCs/NPCs in the RMS adjacent to the cortical infarct area; only bridge NSCs/NPCs remain in the RMS nearby the cortical infarction. b Two migrating NSCs/NPCs physically interact with each other (communication) in the RMS distal to the cortical infarction. c The result of communication: one of the migrating NSCs/NPCs moves forward in the direction of OB (escaping away from the dead zone—cortical infarction), and other migrating NSC/NPC travels towards the infarction area along the RMS. d The migrating NSC/NPC “communicates” with a bridge NSC/NPC in the RMS close to the zone of dying brain cells (the cortical infarction area). e and f The results of the communication shown in d. The migrating NSC/NPC flees away from the dead zone (cortical infarction). The schematic diagram summarizes the results of time-lapse images published elsewhere (Zhao and Nam 2007). LV lateral ventricle. RMS the rostral migratory stream
2.3 The Early Reaction of NSCs/NPCs in Response to Cortical Brain Ischemia is Self-Protective
Time-lapse images revealed that the early response to cortical brain ischemia for NSCs/NPCs was to escape away from the area of brain damage (Zhao et al. 2007a). Cortical brain ischemia was made by the permanent occlusion of the right common carotid artery and the right middle cerebral artery distal to the striatal branch . Brain slices were prepared 3 h after induction of cortical brain ischemia in the right hemisphere of nestin-GFP mice, live brain slice imaging was acquired with a multiphoton microscope, and time-lapse images were recorded for 15 h at 7 min intervals. Very interestingly, we observed that only bridge NSCs/NPCs remained in the RMS next to the infarct cortex, whereas there were no migrating NSCs/NPCs in the RMS adjacent to the infarct cortex (Fig. 2.1 b–f). The migrating NSCs/NPCs were seen in the RMS distal to the infarct area. The migrating NSCs/NPCs in this part of RMS continued touching each other for a while (Fig. 2.1 b); thereafter, some of the NSCs/NPCs immediately moved away from the lesion area and towards to the direction of OB, and some other NSCs/NPCs traveled toward to the direction of infarct area along the RMS (Fig. 2.1 c). However, once they touched the bridge NSCs/NPCs in the location closer to the infarct cortex (Fig. 2.1 d), these migrating NSCs/NPCs changed their direction 180° and started moving away from the infarct area (Fig. 2.1 e and f). This observation suggests that cell-cell communication between the NSCs/NPCs leads to changes of direction for the migrating NSCs/NPCs, and that the early reaction of the migrating NSCs/NPCs in response to ischemic damage in the cortex is to move away from the infarct area during the 3–18 h after the induction of cortical ischemia . The results of this study offer insights into the natural self-protective behavior of migrating NSCs/NPCs in response to brain ischemic injury in the early period of cortical ischemia–-escape from the infarct area, and this behavior is self-protection .
The open questions to be addressed in the future studies are: how the NSCs/NPCs sense the microenvironment changes, how the signals are transported/delivered among the NSCs/NPCs, and how NSCs/NPCs escape away from “danger”– the dead zone (infarct) induced by focal brain ischemia .
2.4 Focal Brain Ischemia-Induced NSC/NPC Proliferation is Required for Brain-Self Protective
It has been well documented that focal brain ischemia is a robust trigger for stimulating NSC/NPC proliferation in both SVZ and SGZ in the brain of adult rodents (Arvidsson et al. 2001; Jin et al. 2001; Arvidsson et al. 2002) . It has been proposed that focal brain ischemia-induced NSC/NPC proliferation and neurogenesis may play a role in brain self-repair. However, more than 80 % of newly formed neurons have been found dead in the setting of focal brain ischemia, and the surviving newborn neurons replace only 0.2 % of dead neurons by ischemic injury (Arvidsson et al. 2002). Therefore, the capability of brain-self repair by the endogenous NSCs/NPCs in the adult brain after ischemic injury is limited. Is brain ischemia-induced NSC/NPC proliferation per se actually involved in brain self-repair/protection? To address this question, we performed experiments in which NSC/NPC proliferation was blocked with a cell proliferating inhibitor, and we then determined whether NSC/NPC proliferation triggered by brain ischemia is vital for brain self-protection .
Cortical brain ischemia was produced by the permanent occlusion or ligation of the right common carotid artery and the middle cerebral artery distal to the striatal branch in male adult C57BL mice or spontaneously hypertensive rats (SHRs). Cytosine-β-D-Arabinofuranoside (Ara-C), a DNA synthesis inhibitor to prevent cell proliferation , was infused into the cerebral lateral ventricle ipsilateral to the ischemic hemisphere with a 7-day-infusion osmotic minipump. Ara-C was initially delivered one day before the induction of cortical brain ischemia or immediately after the induction of cortical brain ischemia, and the infusion of Ara-C was continued for 7 days. Bromodeoxyuridine (BrdU), a synthetic thymidine analog, can be incorporated into DNA during cell dividing. To identify proliferating NSCs/NPCs, BrdU was intraperitoneally injected daily during Ara-C infusion. We found that cortical brain ischemia-induced NSC/NPC proliferation in the bilateral SVZ and ipsilateral SGZ was completely inhibited by Ara-C (Li et al. 2010). In addition, slow dividing type −1 NSCs/NPCs in the bilateral SGZ were also significantly reduced by Ara-C infusion (Li et al. 2010). These findings indicate that cerebral lateral ventricle infusion of Ara-C is sufficient to prevent cortical brain ischemia-induced NSC/NPC proliferation in both SVZ and SGZ .
Our research data also revealed that inhibiting cortical brain ischemia-induced NSC/NPC proliferation in both SVZ and SGZ exaggerated neuron loss in both the hippocampus and infarct cortex) (Li et al. 2010). There are two types of neuron loss in the setting of focal brain ischemia: primary neuron loss and secondary neuron loss. The primary neuron loss occurs in the ischemic core within several hours after focal brain ischemia, whereas the secondary neuron loss appears in the brain tissue remote to the infarct core days and even months after focal brain ischemia (Hara et al. 1993; Touzani et al. 2001). It is worth noting that focal brain ischemia-induced NSC/NPC proliferation in the SVZ and SGZ is markedly increased during the period of 4 days to 2 weeks post-ischemia (Jin et al. 2001; Arvidsson et al. 2002) Therefore, focal brain ischemia-induced amplification of NSCs/NPCs in the SVZ and SGZ may contribute to protecting neurons from the ischemia-induced secondary death. The evidence supporting this hypothesis shows that inhibiting NSC/NPC proliferation in the SGZ causes significant neuron loss in the hippocampus 7 days after cortical brain ischemia (Li et al. 2010). In addition, the neuron loss in the hippocampus has been demonstrated as the secondary neuron loss through apoptosis (Li et al. 2010) because the stroke model used in this study only causes infarction in the ipsilateral cortex, whereas the hippocampus is spared from the infarct .
How focal brain ischemia-induced amplification of NSCs/NPCs in the SVZ and SGZ protects brain from ischemic damage remains poorly understood. One possibility may be due to releasing trophic factors as our findings have shown that adult brain-derived NSCs/NPCs can protect cortical neurons against excitotoxic damage through the NSC/NPC-released trophic factors, brain-derived neurotrophic factor (BDNF) and vascular endothelial growth factor (VEGF) (Li et al. 2010). In agreement with these observations, other investigators have also found that both BDNF and VEGF are produced by NSCs/NPCs (Leker et al. 2007; Mochizuki et al. 2008) and that administration of BDNF and VEGF in animal models of brain ischemia leads to reduction in infarction size (Schabitz et al. 1997; Ren and Finklestein 2005) .
NSCs/NPCs may act as trophic factor providers to save neurons from the injury of focal brain ischemia. Several lines of evidence have shown that the NSCs/NPCs in the SVZ of adult brain can migrate long distances to the cortex adjacent to the infarct area. It has been demonstrated that the neuroblasts derived from proliferating NSCs/NPCs in the SVZ can migrate to the peri-infarct cortex through the white matter (Jin et al. 2003) and blood vessels (Ohab et al. 2006). In line with these findings, we also observed that SVZ-derived NSCs/NPCs migrated toward the cortical infarction along the corpus callosum at day 7 after induction of cortical brain ischemia (Li et al. 2010). Proliferating NSCs/NPCs in the SGZ may rescue hippocampal neurons through the delivery of BDNF via axonal projections between the dentate gyrus and hippocampal CA1 and CA3 subregions. It has been revealed that trkB, the receptor for BDNF, is expressed in CA1 and CA3 (Yan et al. 1997). BDNF has been shown to prevent glutamate-induced apoptotic cell death (Almeida et al. 2005) .
Our knowledge and understanding of the precise role of focal brain ischemia-induced amplification of NSCs/NPCs in the SVZ and SGZ in adult brain is still far from complete. Is the focal brain ischemia-induced amplification of NSCs/NPCs part of a reaction from NSCs/NPCs to protect themselves in the condition of ischemic injury? It has been shown that a widespread hyperexcitability occurs in the intact brain regions including peri-infarct cortex and hippocampus , and that the hyperexcitability is most pronounced at 3–7 days and persists 1 month after cerebral cortical ischemia (Schiene et al. 1996; Qu et al. 1998; Redecker et al. 2002). NSCs/NPCs in the adult brain share many physiological features with astrocytes (Filippov et al. 2003; Kempermann et al. 2004b). Astrocytes can regulate inhibitory synapses formation in hippocampal neurons (Elmariah et al. 2005), manipulate glutamatergic transmission and uptake glutamate (Newman 2003). Can focal brain ischemia-induced amplification of NSCs/NPCs in both SVZ and SGZ contribute to preventing the hyperexcitability-induced neuron damage ?
Other interesting questions that remain elusive are: whether generating and releasing trophic factors from NSCs/NPCs are cell-status dependent (proliferation vs. quiescent), and whether there are other mechanisms involved in NSC/NPC -induced neuroprotection in addition to the trophic factors released by NSCs/NPCs. Recent studies have revealed that exosomes and microvesicles (EMVs) play a key role in cell-to-cell communication. EMVs contain membrane receptors, proteins, mRNAs, miroRNAs and organelles that offer genetic regulation of cell survival, phenotype, proliferation , differentiation or death to the target cells either nearby (paracrine) or at a distance (endocrine) (Camussi et al. 2010). Stem cells have been demonstrated to release abundant EMVs (Camussi et al. 2010; Aoki et al. 2014). Stem cell-derived EMVs have been shown to rescue cells from injury, reprogram terminally differentiated cells to re-enter the cycle, and promote tissue regeneration and repair (Camussi et al. 2010). However, the role of NSC/NPC-released EMVs in supporting brain tissue survival and regeneration in the setting of stroke remains an open question to be addressed in future .
Enhancing propagation of NSCs/NPCs has been shown to protect brain from ischemic injury in animal models. Systemic administration of stem cell factor (SCF) 3h after induction of cortical brain ischemia in SHRs dramatically increases NSC/NPC proliferation in the SVZ, reduces infarction size and eliminates neurological deficits (Zhao et al. 2007a). Sodium butyrate has been reported to enhance amplification of NSCs/NPCs, reduce infarction size and improves functional outcome when systemically administered for 7 days after focal brain ischemia in rats (Kim et al. 2007; Kim et al. 2009). Leker and co-workers (2007) (Leker et al. 2007) demonstrated that long-term delivery of fibroblast growth factor-2 (bFGF) immediately after induction of cortical brain ischemia in SHRs resulted in the enhancement of NSC/NPC proliferation in the SVZ, white matter (the corpus callosum) and peri-infarct cortex throughout 90 days after ischemia and the improvement of motor function. These findings provide insights into developing new therapeutic approaches for brain protection through increasing endogenous NSCs/NPCs .
2.5 Hematopoietic Growth Factors Govern NSC/NPC Lineage Differentiation
Stem cell factor (SCF) and granulocyte-colony stimulating factor (G-CSF) were initially discovered as the essential hematopoietic growth factors that regulate the mobilization, proliferation and differentiation of hematopoietic stem cells/hematopoietic progenitor cells (HSCs/HPCs) (Ashman 1999) (Ripa and Kastrup 2008). Accumulating evidence has revealed that these hematopoietic growth factors may also play roles in the central nervous system (CNS). SCF (Zhao et al. 2007a) and G-CSF (Schneider et al. 2005; Zhao et al. 2007a) have been proven to pass through the blood-brain barrier in intact rodent brains. Systemic administration of SCF (Zhao et al. 2007b; Zhao et al. 2007a) and G-CSF (Schabitz et al. 2003; Six et al. 2003; Shyu et al. 2004; Schneider et al. 2005; Komine-Kobayashi et al. 2006; Zhao et al. 2007a) alone, or SCF in combination with G-CSF (Zhao et al. 2007b; Zhao et al. 2007a) has been shown to protect the brain against ischemic injury (Schabitz et al. 2003; Six et al. 2003; Shyu et al. 2004; Schneider et al. 2005; Komine-Kobayashi et al. 2006; Zhao et al. 2007a) and facilitate brain repair during chronic stroke (Zhao et al. 2007b). In addition, SCF and G-CSF appear to be involved in neuronal plasticity. Mice with mutations of SCF (Motro et al. 1996) or SCF receptor (Katafuchi et al. 2000) show impaired long-term potentiation (LTP) and spatial learning and memory. G-CSF deficient mice display cognitive impairment, LTP reduction, and poor neuronal networks in the hippocampus (Diederich et al. 2009). Interestingly, SCF receptor, ckit, and G-CSF receptor, GCSFR, have been found to be expressed in the NSCs/NPCs of the adult rodent brain (Schneider et al. 2005; Zhao et al. 2007a). However, the effects of SCF and G-CSF on the NSCs/NPCs remain largely unknown.
Using the approaches of cellular biology and molecular biology, we have determined the regulatory effects of SCF and G-CSF on lineage commitment of NSCs/NPCs. First of all, we observed that the receptors for SCF and G-CSF were expressed on the NSCs/NPCs in the ventricular zone of the developing rat brain at embryonic day 18 (Piao et al. 2012). The NSCs/NPCs were then isolated from the ventricular zone of the embryonic rat brain at E18, a time period in which both neurogenesis and gliogenesis take place. The NSCs/NPCs were then grown in cell culture dishes to form neurospheres. After forming the secondary neurospheres, NSCs/NPCs were disassociated into single cells and were then allowed to differentiate in a differentiation medium. SCF and G-CSF alone or in combination were added at the beginning of the differentiation stage of NSCs/NPCs. We found that SCF and G-CSF alone or in combination increased the number of TuJ1-positive neuronal cells and reduced GFAP-positive astrocytes (Piao et al. 2012), suggesting a regulative role of SCF and G-CSF on lineage switch of NSCs/NPCs. In line with our observation, other investigators have revealed that SCF (Jin et al. 2002) and G-CSF (Schneider et al. 2005), promote neurogenesis in vivo.
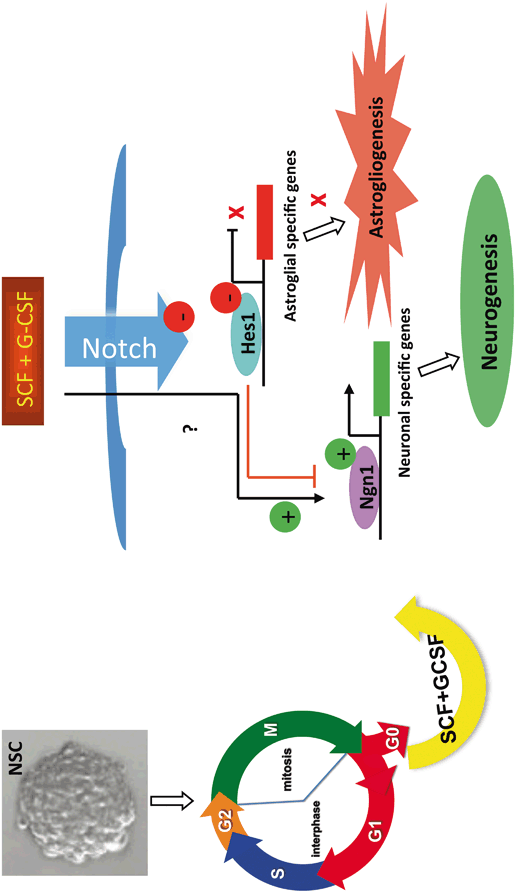
Very interestingly, SCF and G-CSF can promote neuronal fate determination of NSCs/NPCs at the proliferation stage. SCF and G-CSF alone or in combination were added to the dividing NSCs/NPCs in the secondary neurospheres for 3–4 days. Thereafter, the mitogens (bFGF and EGF) and the hematopoietic growth factors were then withdrawn, and the NSCs/NPCs were allowed to differentiate. We observed that only SCF in combination with G-CSF (SCF+G-CSF) treatment showed a significant increase in neurogenesis and reduction in gliogenesis (Piao et al. 2012). This observation led us to hypothesize that SCF+G-CSF promotes neuronal lineage commitment of NSCs/NPCs through the regulation of the cell cycle. To test this hypothesis, we examined the effects of SCF+G-CSF on the cell cycle and NSC/NPC propagation. The results of a cell cycle assay showed that SCF+G-CSF increased the population of NSCs/NPCs at the G1/G0 phase and decreased the population of NSCs/NPCs at the S phase (Piao et al. 2012), suggesting that SCF+G-CSF causes the cell cycle arrest of NSCs/NPCs. To further confirm this result, BrdU was added into the NSCs/NPCs during the proliferating stage. We chose BrdU for labeling the dividing NSCs/NPCs at the S phase because BrdU is a thymidine analogue and can be incorporated into the newly synthesized DNA in S-phase cells. We found that SCF+G-CSF treatment during NSC/NPC proliferation resulted in a significant reduction of BrdU positive NSCs/NPCs, indicating that the NSC/NPC dividing is inhibited by SCF+G-CSF (Piao et al. 2012). Furthermore, when we examined NSC/NPC growth curve, we observed that NSC/NPC propagation was reduced by SCF+G-CSF (Piao et al. 2012). Together, these data suggest that SCF+G-CSF leads to cell cycle exit of NSCs/NPCs and inhibits NSC/NPC proliferation. Inducing cell cycle arrest of NSCs/NPCs is the critical and initial step to promote neuronal lineage differentiation of NSCs/NPCs . This notion is supported by the findings that pro-differentiation molecule-induced neuronal differentiation is preceded by cell cycle withdrawal (Henrique et al. 1997; Farah et al. 2000; Politis et al. 2008) and that promoting the cell cycle leads to inhibition of neuronal differentiation (Ohnuma et al. 2001).
How does SCF+G-CSF induce neuronal fate determination and commitment of NSCs/NPCs? To address this question, we examined the effects of SCF+G-CSF on the regulation of proneural bHLH transcription factors and Notch signaling pathway. Convincing evidence has shown that neurogenin 1 (Ngn1), one of the proneural bHLH transcription factors, promotes neuronal differentiation of NSCs/NPCs . Ngn1 has been found to be expressed in the ventricular zone, and Ngn1 only appears during cortical neurogenesis (Gradwohl et al. 1996; Sommer et al. 1996; Ma et al. 1997). In fact, Ngn1 acts as an activator of neuronal transcription factor that promotes neurogenesis and inhibits glial differentiation (Sun et al. 2001). Erythropoietin, one of the hematopoietic growth factors , has been shown to promote neuronal differentiation of NSCs/NPCs through Ngn1 mediation (Wang et al. 2006). By contrast, Notch1/Hes1 signaling contributes to maintaining NSCs/NPCs in an undifferentiated stage (Artavanis-Tsakonas et al. 1999; Mizutani and Saito 2005), enhances astroglial differentiation (Schmid et al. 2003; Anthony et al. 2005), and suppresses neuronal lineage commitment of NSCs/NPCs (Kageyama and Ohtsuka 1999). Based on these findings, we hypothesized that SCF+G-CSF promotes neuronal differentiation of NSCs/NPCs through acting the Ngn1 transcription and inhibiting the Notch1/Hes1 signaling. To test this hypothesis, we quantified gene expression of NSCs/NPCs with real-time RT-PCR after SCF+G-CSF treatment. The gene expression data showed that Ngn1 gene expression was significantly up-regulated by SCF+G-CSF. In addition, both Notch1 and Hes1 genes were significantly down-regulated by SCF+G-CSF (Piao et al. 2012). Moreover, when Ngn1 transcription was silenced by the siRNAs against Ngn1, SCF+G-CSF-induced enhancement of neuronal lineage commitment and inhibition of astroglial differentiation was significantly prevented (Piao et al. 2012). These findings suggest that Ngn1 is required for SCF+G-CSF-induced lineage determination and neuronal fate commitment of NSCs/NPCs. A putative model of the effects of SCF+G-CSF on cell cycle withdrawal and cell fate switch in NSCs/NPCs is presented in Fig. 2.2.
Related posts:
 The Role of Endogenous Neural Stem Cells in Stroke
Bone Marrow Mesenchymal Stromal Cell Transplantation: A Neurorestorative Therapy for Stroke
Basic Studies on Neural Stem Cells in the Brain
Transplantation of Adipose-Derived Stem Cells in Stroke
Adipose-Derived Stem Cells: Isolation and Culturing
The Contribution of Mesenchymal Stromal Cells in Traumatic Brain Injury
The Role of Endogenous Neural Stem Cells in Stroke
Bone Marrow Mesenchymal Stromal Cell Transplantation: A Neurorestorative Therapy for Stroke
Basic Studies on Neural Stem Cells in the Brain
Transplantation of Adipose-Derived Stem Cells in Stroke
Adipose-Derived Stem Cells: Isolation and Culturing
The Contribution of Mesenchymal Stromal Cells in Traumatic Brain Injury
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


