CHAPTER 27
Neuro-oncology and Transplant Neurology
I. Central Nervous System (CNS) Tumors
A. Epidemiology
1. Metastatic brain tumors outnumber primary brain tumors 10 to 1.
2. Gliomas are the most common primary brain tumors in adults (70%), whereas primitive neuroectodermal tumors (PNETs) are the most common primary brain tumors in children.
3. Genetics and radiation exposure to the head are important risk factors for gliomas.
4. Young age, high performance status, and low pathological grade are favorable prognostic factors for primary brain tumors.
5. Children less than 1 year old (y/o) have mainly supratentorial tumors; after 1 year, approximately 70% are infratentorial; only 30% of tumors in adults are infratentorial.
6. The most common infratentorial pediatric brain tumors include medulloblastoma, cerebellar astrocytoma, brainstem glioma, and ependymomas.
7. The most common supratentorial pediatric tumors are gliomas and craniopharyngioma.
8. Common differentials for intracranial masses include CNS infections, atypical stroke/vasculitis, tumefactive multiple sclerosis (MS), and radiation necrosis.
B. Oncogenes and chromosomal aberrations in the CNS (see Chapter 2)
C. Neuroepithelial tumors
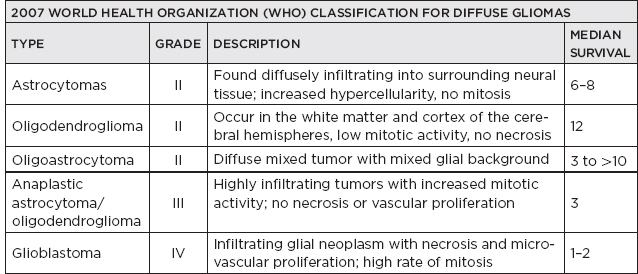
II. Common CNS/Peripheral Nervous System (PNS) Tumors
TUMOR TYPE | COMMENTS/CLINICAL FEATURES | PATHOLOGY |
Astrocytic tumors | ||
Fibrillary | Grade II; age 30–50 years Malignant change after several years occurs frequently; complete resection/cure often not possible Do not enhance on scans | Sparse and fairly regular proliferation of astrocytes without histologic evidence of malignancy, mitotic figures, or vascular endothelial proliferation and without area of necrosis or hemorrhage |
Anaplastic | Considered Grade III; poorly responsive to therapy; frequently evolve into glioblastoma | Cellular atypia, mitotic activity and endothelial proliferation is common (but no necrosis) |
Glioblastoma | Most malignant grade; also the most common malignant primary tumor in adults (50% of all gliomas); chiefly supratentorial; MRI: enhancing lesion surrounded by edema (may be indistinguishable from abscess); frequently crosses the corpus callosum (butterfly glioma) | Pathologically demonstrate a pseudopalisading pattern (arrangement of nuclei around an area of necrosis) |
Juvenile pilocytic astrocytoma (JPA) | More common in children and young adults; location: commonly in the cerebellum, optic nerve, hypothalamus; generally discrete and well circumscribed MRI: cystic mass with enhancing mural nodule; other differential for intracranial lesions presenting with this imaging include pleomorphic xanthoastrocytoma (PXA) and ganglioglioma. 80% 20-year survival | Cystic structures containing mural nodule, hair-like cells with pleomorphic nuclei, contain Rosenthal fibers (opaque, homogeneous, eosinophilic structures), microcystic, endothelial proliferation |
Subependymal giant cell astrocytoma (SEGA) | Grade I astrocytoma; well demarcated; location: lateral ventricles, may cause obstructive hydrocephalus; associated with tuberous sclerosis; may produce hydrocephalus if it occurs at the foramen of Monro; MRI: well circumscribed, enhance homogenously | Large, round nuclei, few mitoses, few endothelial proliferations; prognosis: good |
Pleomorphic xanthoastrocytoma (PXA) | Common in adolescents and young adults; located in the superficial cortex; frequently in the temporal lobe; thus, often causes seizures Good prognosis | Often cystic, large pleomorphic, hyperchromatic nuclei, minimal mitotic activity, no necrosis Eosinophilic granular bodies usually seen; Rosenthal fibers rare |
Gemistocytic astrocytoma | Variant of astrocytoma 80% subsequently transform into glioblastoma | Contains neoplastic astrocytes with abundant cytoplasm |
Gliomatosis cerebri | Fairly uncommon; hemispheric or bihemispheric, poor prognosis | Diffuse, neoplastic astrocytic infiltration |
Gliosarcoma | Prognosis and treatment are similar to glioblastoma | Collision tumor; combination of malignant glial and mesenchymal elements |
Ependymal tumors | ||
Ependymoma | Account for 6% of gliomas; occur at any age but more common in childhood and adolescence; location: more likely infratentorial, with the most frequent site being the 4th ventricle; in the spinal cord, 60% are located in the lumbosacral segments and filum terminale Prognosis has improved, although tumor generally recurs at some point | Gross—reddish nodular and lobulated; microscopic—rudimentary canal and gliovascular (pseudo) rosettes, ciliated cells, but can have Flexner-Wintersteiner (true) rosettes; glial fibrillary acidic protein (GFAP positive) |
Anaplastic ependymomas | Usually well circumscribed and benign |
|
Myxopapillary ependymomas | More commonly at the cauda equina or filum terminale; may mimic herniated disc or present with posterior rectal mass; generally benign and well circumscribed Differential for filum terminale tumor is paraganglioma | Contain mucinous structures surrounded by ependymal cells GFAP and S-100 positive |
Subependymoma | Usually incidental findings at autopsy in adults; well-circumscribed ventricular lesions; usually asymptomatic (or can cause obstructive hydrocephalus) | Cluster of nuclei separated by acellular areas; GFAP positive |
Choroid plexus tumors | ||
Choroid plexus papilloma | Account for 2% of intracranial tumors; most frequently in the 1st decade of life; location: 4th ventricle, lateral ventricle (left > right), 3rd ventricle; in children, more commonly supratentorial; in adults, more commonly infratentorial; more common to cause ventricular obstruction than cerebrospinal fluid (CSF) overproduction; MRI: well-delineated ventricular lesion with fairly homogeneous enhancement; Curable by surgical resection; does not invade the brain (no recurrence) | Papillary, calcified, single layer of cuboidal or columnar cells with a fibrovascular core |
Choroid plexus carcinoma | Also affects young children; location: 4th ventricle, occipital lobe | Less differentiated, increased mitosis, necrosis, invades the parenchyma, may seed CSF |
Oligodendroglial tumors | ||
Oligodendroglioma | Account for approximately 5% of intracranial gliomas; most frequently between ages 30 and 50 years; location: usually cerebral hemispheres, clinical behavior is unpredictable and depends on degree of mitotic rate | Pathology: uniform fried-egg cells, delicate vessels, calcification; mitotic figures are rare, GFAP negative; MRI: diffuse wispy white-matter appearance; treatment: radiation and chemosensitive; better prognosis than astrocytic tumors Deletion of 1p19q with better treatment response |
Anaplastic oligodendroglioma | Higher grade with propensity to turn into glioblastoma | Larger pleomorphic nuclei; with mitotic activity, endothelial proliferation and necrosis |
Mixed oligoastrocytomas | Glioblastoma with presence of oligodendroglial component may have better prognosis (more responsive to chemotherapy) | With oligodendroglial and astrocytic components; low grade or anaplastic |
Neuronal and mixed neuronal-glial tumors | ||
Gangliocytoma | Benign tumor most frequently at the floor of the third ventricle followed by the temporal lobe, cerebellum, parieto-occipital region, frontal lobe, and spinal cord | Tumor of mature-appearing neoplastic neurons only |
Ganglioglioma | Mature neoplastic neurons and neoplastic astrocytes; almost always benign; commonly in the temporal lobe of a young adult; may cause long-standing seizure | May appear cystic with a mural nodule, microscopic—binucleate, round, GFAP negative, and synaptophysin-positive neoplastic neurons |
Central neurocytoma | Relatively uncommon intraventricular tumor in the lateral ventricle; discrete, well circumscribed; enhance with contrast administration | Microscopically indistinguishable from oligodendroglioma; small, round, bland nuclei, Homer-Wright rosettes, no histologic anaplasia (benign), confirmation comes with immunohistochemical demonstration of neural antigens or electron microscopy showing neuronal features |
Dysembryoplastic neuroectodermal tumor | Multiple intracortical lesions mainly in the temporal lobe | Small islands of mature neurons mimicking oligodendrocytes floating in a mucin-like substance |
Dysplastic infantile ganglioglioma | Highly characteristic supratentorial neuroepithelial neoplasms that occur as large cystic masses in early infancy; most frequently in the frontal and parietal region Very favorable clinical course after successful complete or subtotal resection | Marked fibroblastic and desmoplastic component but mature neuroepithelial cells of both glial and neuronal lineage; more primitive cells are also present |
Dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos) | Slowly evolving lesion that forms a mass in the cerebellum | Composed of granule, Purkinje, and glial cells; lack of growth potential; therefore, favorable prognosis |
Paraganglioma (chemodectoma) | Neural crest derived; location: filum terminale, supra-adrenal, carotid body; glomus jugulare, glomus vagale; can produce neurotransmitters Treatment: surgery or chemotherapy/radiotherapy | Nodules surrounded by reticulin—Zellballen |
Olfactory neuroblastoma (esthesioneuroblastoma) | Nasal obstruction/epistaxis may be the presenting symptoms; may erode through the cribriform plate | Small blue cell tumor (also a type of primitive neuroectodermal tumor [PNET]); treatment: responsive to chemotherapy/radiotherapy; fairly good prognosis |
Embryonal tumors | ||
PNETs | Small blue cell tumors that are named depending on their location; 50% are calcified; 50% are cystic; propensity to spread along CSF; generally sensitive to radiotherapy (NB: blastomas are PNET tumors except for hemangioblastoma) | They resemble germinal matrix; >90% are nondifferentiated cells (capable of differentiating along astrocytic, ependymal, oligodendroglial, and neuronal lines) |
Medulloblastoma | The most common intracranial PNET; in children, most commonly located at the cerebellar midline (25% of pediatric brain tumors); 50% have drop metastasis; 50% 10-year survival with multimodality therapy (surgery, radiation, chemotherapy) Suspect medulloblastoma in child presenting with headaches and ataxia; loss of alleles on chromosome 17p; oncogenes called c-myc and n-myc are known to be amplified in some medulloblastomas—poor prognostic indicators | Homer-Wright rosettes, carrot-shaped nuclei, mitosis, necrosis |
Pineoblastoma | PNET of the pineal gland; occurs most commonly in children; contrast-enhancing pineal region mass; Treatment: surgical resection; Prognosis: rapid recurrence with wide dissemination | Resembles medulloblastomas, may form fleurettes |
Retinoblastoma | Sporadic in 60% of all cases, autosomal dominant in 40%; emergence of tumor requires inactivation of Rb gene (tumor suppressor gene) on chromosome 13q14; in the familial form, one gene is inactivated in the cell; thus, only one gene needs inactivation to produce the tumor; in the sporadic form, both need inactivation; trilateral (rare): bilateral retinoblastomas + pineoblastoma Treatment: enucleation, intra-arterial chemotherapy | Flexner-Wintersteiner rosettes with mitosis and necrosis |
Neuroblastoma | Rare in the CNS; most commonly arise in the sympathetic chain (or in adrenal) in childhood; clinical correlate of dancing eyes and dancing feet syndrome (opsoclonus myoclonus) Anti-Ri (ANNA-2) antibodies reported in children with neuroblastoma Treatment: ACTH | Small blue cell tumors, Homer-Wright rosettes |
Tumors of nerve sheath cells | ||
Neurilemmoma (schwannomas; neurinoma) | Benign tumors arising from Schwann cell; usually solitary except in neurofibromatosis type 1, in which they are multiple; in neurofibromatosis type 2, bilateral acoustic schwannomas can be found; location: most commonly cranial nerve VIII, also other cranial nerves, spinal roots (thoracic segments > cervical, lumbar, cauda equina); MRI: hyperintense on T2; gross total resection usually possible; Schwannomas are strongly positive for S-100 protein | Antoni type A—dense fibrillary tissue, narrow elongated bipolar cells with very little cytoplasm and nuclei that are arranged in whorls or palisades Antoni type B—loose reticulated type tissue, round nuclei are randomly arranged in a matrix that appears finely honeycombed |
Neurofibroma | Differ from schwannomas in that they almost always occur within the context of neurofibromatosis type 1, almost always multiple, may undergo malignant transformation in 0.5%–1.0% of tumors (neurofibrosarcoma); plexiform neurofibroma: cord-like enlargement of nerve twigs | Single cells, axons, myxoid background, parent nerve is usually intermingled with tumor |
Tumors of the meninges | ||
Meningioma | Benign tumors originating from arachnoid cells; account for 13%–18% of primary intracranial tumors and 35% of intraspinal tumors (frequently in the thoracic segment in the lateral compartment of the subdural space); most meningiomas have a partial or complete deletion of chromosome 22; primarily in adults (20–60 y/o), although it may occur in childhood; female predominance, especially in the spinal epidural space (NB: women with breast cancer have a higher incidence of meningioma) Radiology: dural-based tumors cause enhancement of the peripheral rim of the dura surrounding the meningioma, producing the dural tail Location: convexity meningiomas (parasagittal, falx, lateral convexity); basal meningiomas (olfactory groove, lesser wing of the sphenoid, pterion, suprasellar meningiomas); posterior fossa meningiomas, meningiomas of the foramen magnum, as well as intraventricular meningiomas, are considerably less common Foster Kennedy syndrome, characterized by contralateral papilledema and ipsilateral optic atrophy and anosmia, can occur when a meningioma directly compresses ipsilateral optic nerve in olfactory groove | Gross—spherical, well circumscribed, and firmly attached to the inner surface of the dura; microscopic—psammoma bodies (whorls of cells wrapped around each other with a calcified center), xanthomatous changes (presence of fat-filled cells), myxomatous changes (homogeneous stroma separating individual cells), areas of cartilage or bone within the tumor, foci of melanin pigment in the connective tissue trabeculae, rich vascularization; may produce hyperostosis (local osteoblastic proliferation of skull); most are epithelial membrane antigen positive |
Hemangiopericytoma | Tumors originating from fibroblasts in the dura Considered a variant of solitary fibrous tumor | Irregular, elongated tumor cells with a mesenchymal component Stain strongly for CD34 |
Melanocytoma | Most commonly posterior fossa tumors arising from meningeal melanocytes, usually slow growing and low grade Important to distinguish from malignant melanomas, which are aggressive with a poor 5-year survival | Melanocytomas are usually spindle shaped or fusiform, looking much like meningiomas If associated necrosis and hemorrhage is seen, consider melanoma. |
Lipoma | Low-grade tumors, often inter-hemispheric/pericallosal, with very slow growth Arise from mesenchymal component within meninges May be associated with vascular malformations and present with headaches or seizures | Mature adipose tissue containing engorged blood vessels; often found in subarachnoid space |
Tumors of uncertain histogenesis | ||
Hemangioblastoma | The most common primary cerebellar neoplasm; sporadic or genetic; 20% are associated with von Hippel-Lindau syndrome (hemangioblastoma, retinal angiomatosis, renal cell carcinoma, renal and pancreatic cysts); location: cerebellum > brain stem > spinal cord Gadolinium-enhanced MRI is the best modality for detection | Gross—cystic lesion with mural nodule; microscopic—foamy cells in clusters separated by blood-filled channels, surrounding parenchyma contains Rosenthal fibers |
Lymphomas and hematopoietic neoplasms | ||
Malignant lymphoma | Most CNS lymphomas are B cell; usually non-Hodgkin’s (usually a diffuse large cell variety); more common in the immunocompromised population; Epstein-Barr virus may play a role; ghost tumor: initially may respond dramatically to steroids and/or radiation but eventually recurs; commonly originate in the basal ganglia or periventricular white matter; may be radiology: homogeneous contrast enhancement located in the deep brain rather than the gray–white interface; Treatment: methotrexate; responds to steroids and radiation but recurs Prognosis: overall survival in HIV is 3 months or less, in non-HIV is 19 months | Multifocal; angiocentric or perivascular distribution |
Cysts and tumor-like lesions | ||
Rathke cleft cyst | Epithelial cyst in the sella |
|
Epidermoid cyst | Radiology: low-density cyst with irregularly enhancing rim, may not enhance with contrast; Treatment: surgical excision preferred Prognosis: recurrence with subtotal excision | Occur due to slow-growing ectodermal inclusion cysts; secondary to ectoderm trapped at the time of closure of neural tube; cyst lined by keratin-producing squamous epithelium; leakage of contents into the CSF produces chemical meningitis; occurs mostly laterally instead of midline |
Dermoid cyst | Mostly present in childhood; hydrocephalus common Location: usually midline, related to fontanel, 4th ventricle, spinal cord; Treatment: surgical resection Prognosis: recurrence with subtotal excision | Comprising mesoderm and ectoderm lined with stratified squamous epithelium and filled with hair, sebaceous glands, and sweat glands |
Colloid cyst | Account for 2% of intracranial gliomas; mainly in young adults; location: always at the anterior end of the 3rd ventricle, adjacent to the foramen of Monro; MRI: increased signal on T1-weighted images (due to the proteinaceous composition of contents); obstructive hydrocephalus is common Therapy: drainage, surgical resection | |
Hypothalamic hamartomas | Rare; associated with gelastic seizures and endocrine abnormalities; location: hypothalamus; radiology: small discrete mass near the floor of the 3rd ventricle Treatment: surgical resection or ablation, if possible Prognosis: cure is possible if resected | Well-differentiated but disorganized neuroglial tissue |
Tumors of the sellar region | ||
Pituitary adenoma | 15% of all intracranial neoplasms; women > men; most common neoplasm of the pituitary gland; can present early if they hypersecrete hormones or later owing to compressive effects; microadenoma (<10-mm diameter) tend to be hormone secreting, with hyperprolactinemia as the most common hormonal abnormality; may be due to hypersecretion or stalk effect in which the flow of prolactin inhibitory factor (dopamine) is absent; Treatment: bromocriptine—may induce tumor fibrosis (which may make pathologic diagnosis difficult) | a. Acidophilic: growth hormone ± prolactin; rare follicle-stimulating hormone or luteinizing hormone; acromegaly b. Basophilic:adrenocorticotropic hormone >> thyroid-stimulating hormone; hyperadrenalism (Cushing’s disease) c. Chromophobic: prolactin; null; rarely follicle-stimulating hormone/luteinizing hormone; amenorrhea-galactorrhea; men impotent d. Mixed |
Craniopharyngioma | Rathke pouch cyst; origin is uncertain; bimodal age of distribution of childhood and adult life | Calcified and cystic; crankcase oil; more commonly adenomatous but can be papillary; benign but locally adherent |
Germ cell tumors | Occur in midline, mostly in first three decades; much more common in children; most common germ cell tumor is Germinoma; prognosis is good | Histologically similar to other germ cell tumors such as ovarian or testicular; may be mixed Reactive to alpha fetoprotein, embryonal carcinoma, placental alkaline phosphatase, HCG, CD30 |
Pineal gland tumors | ||
Pineocytoma | WHO grade I, symptoms due to local compression of adjacent structures—tectum compression can cause Parinaud’s syndrome (up-gaze paralysis and convergence nystagmus) | Well circumscribed, slow-growing tumor composed of pinealocyte-like cells Small, well-differentiated cells with oval nuclei. Normal pineal gland tissue is lobular and contains calcifications |
Pineoblastoma | Aggressive tumor primarily affecting children; WHO grade IV; 40% of all pineal parenchymal tumors; can be associated with retinoblastoma | Densely packed small blue cells; irregular nuclei with necrosis |
Metastatic tumors | 20–40% of all brain tumors; well defined, round, with surrounding edema; location: gray–white junction; most common site of CNS metastasis is cerebellum. 1. Primary CNS tumors: can metastasize to extracranial regions (any anaplastic glial tumor, PNET, meningioma), lymph nodes, and/or lung (gliomas, meningiomas) or bone (PNET) 2. Secondary metastasis to brain: bronchogenic cancer > breast cancer > melanoma > hypernephroma 3. Hemorrhagic transformation—melanoma, bronchogenic cancer, choriocarcinoma (the only pineal region tumor that may bleed spontaneously; increased β-human chorionic gonadotropin), renal cancer, thyroid cancer 4. Metastasis to the skull/dura: prostate, lung, breast, lymphoma 5. Meningeal carcinomatosis: adenocarcinomas (gastrointestinal, breast, lung) | |
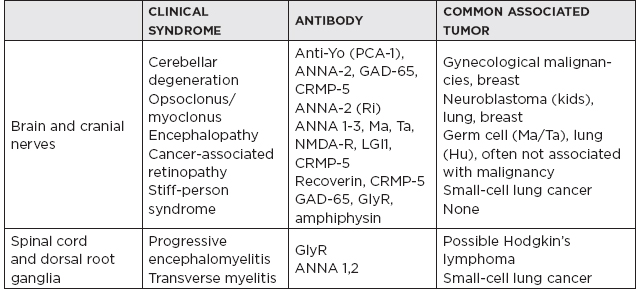
III. Paraneoplastic Syndromes
A. Group of disorders associated with tumors but not directly caused by tumor invasion. They are likely due to autoimmune mechanism affecting neural and nonneural tissue. Neurologically, these immune phenomena may target:
1. Neuromuscular junction (e.g., myasthenia gravis)
2. Target neurons in central or peripheral nervous system (e.g., anti-Hu Ab)
3. Intracellular proteins (e.g., GAD 65)
4. Surface antibodies (e.g., NMDA-R)
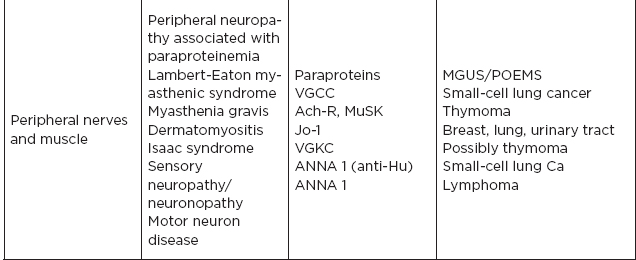
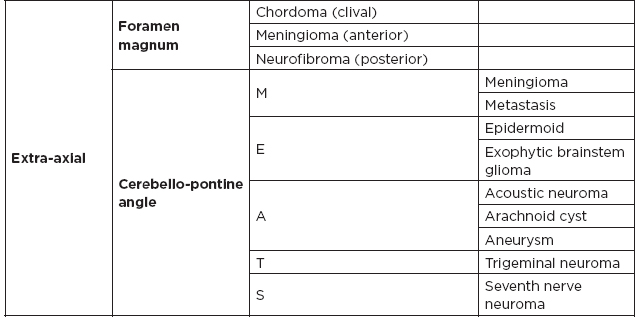
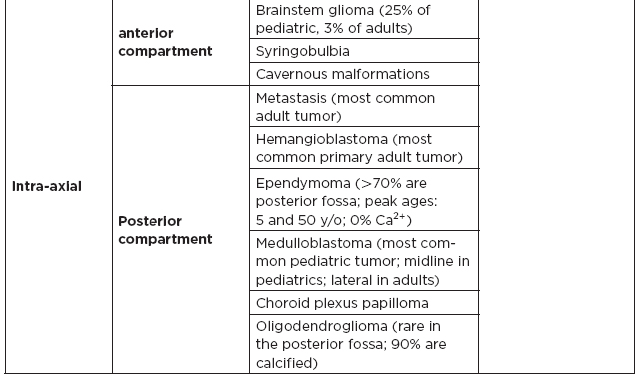
IV. Common Tumor Classifications
A. Most common tumors by location
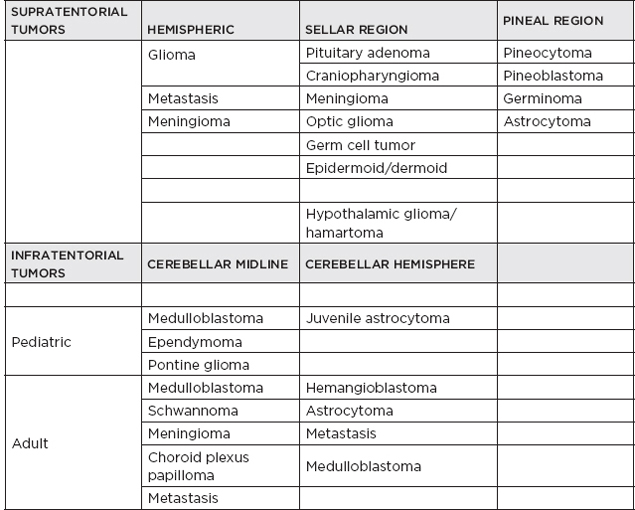
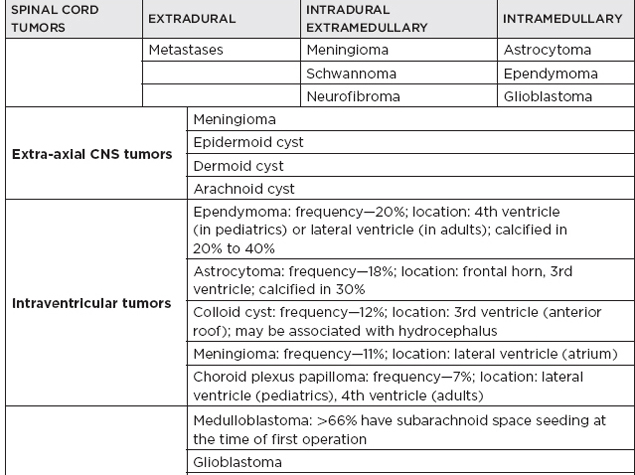

B. Posterior fossa lesions