Fig. 4.1
The fear network in PTSD: sensory (rectangles), emotional (circles), cognitive (thought bubbles), and interoceptive (triangles) elements of the network are highly interconnected, as opposed to the autobiographical context information (rectangles below) which is stored separately (From Wilker and Kolassa (2013), reprinted with permission)
Evidence not only from cognitive psychology but also from neuroscience confirms distinct neural bases to the abstract, flexible, contextualized representations (cold memory) and to the inflexible, sensory-bound representations (Brewin et al. 2010; Kolassa and Elbert 2007). The latter, part of the hot memory system, is thought to be supported primarily by areas of the brain directly involved in perception (e.g., representational cortex, amygdala, insula) rather than in higher-order cognitive control. By contrast, cold memories require the involvement of the hippocampus and surrounding medial temporal lobe structures (Brewin et al. 1996, 2010). For emotional experiences within the normal range, hot and cold information are well integrated and stored together in autobiographical memory. Yet, if PTSD develops after trauma exposure, these two forms of memory become dissociated for two reasons: first, the extreme state of emotional arousal during a traumatic event, accompanied by the release of stress hormones, affects hippocampal functioning, which could explain the reduced consolidation of declarative memories in PTSD (Elzinga and Bremner 2002). Second, the experience of several traumatic experiences, which generally precedes the onset of PTSD, leads to the activation of the same fear memory structure, since different traumatic experiences share common elements (e.g., fear, screams, and blood). Hence, the hot memories for several traumatic experiences merge in one fear network and strengthen the associative interconnections of its elements. This makes it increasingly difficult to disentangle the different traumatic experiences and, particularly, to recall the adequate corresponding context information. Following principles of associative learning, an interconnected fear network thus encompasses sensory, cognitive, physiological, and emotional experiences and includes the action disposition related to the experience (sensory representation, hot memory) but is detached from the autobiographical context information. Furthermore, the strength of the interconnections of the fear network, as well as the number of elements integrated in the network, increases with accumulating trauma exposure, which explains the aforementioned dose-response effect of traumatic load. Environmental stimuli (e.g., a smell or noise) and internal cues (e.g., a thought), usually in combination, can later activate this network at any given time. The ignition of only a few elements in the network is sufficient for activating the whole structure and leads to strong involuntary memories (intrusive symptoms). This may express itself as a flashback, a perception that one is back in the traumatic situation with its sounds of the harsh voice, smells of blood, feelings of fear, and thoughts of dying (cf. Fig. 4.1). Since the activation of the fear network serves as a frightening and painful recollection, many people suffering from trauma-related experiences learn to avoid cues that act as reminders of the traumatic event. They try to avoid thinking about any part represented in the fear/trauma network, not to talk about it, and to stay away from people and places that remind them of the frightening event.
The fear network model corresponds well with results of structural and functional alterations in the brain of trauma survivors with PTSD (see Sect. 4.4 in this chapter) and provides not only a neurobiological explanation of the development of PTSD symptoms in the aftermath of traumatic stress but also a rationale for trauma-focused therapy approaches (Kolassa and Elbert 2007). The reactivation of consolidated memories turns them into a labile state in which modification is possible (Nadel et al. 2012). Hence, the activation of the fear network during treatment allows the modification of the present memory structure through habituation and extinction learning (Ehlers et al. 2010). One example of such an exposure-based treatment is Narrative Exposure Therapy (NET; Schauer et al. 2011 see also Chap. 12 of this book) – a short-term treatment for traumatic stress disorders. By connecting the “hot” sensory, emotional, cognitive, and interoceptive memories of the trauma to the “cold” autobiographical memories, an activation of the fear network disconnected from context can be prevented and the client can achieve control over the distressing memories. The therapist further supports the client to work through the experienced traumatic experiences in a chronological order, which helps to disentangle the traumatic events which merge in the fear network. The effectiveness of NET supports the concept of a fear network in PTSD (see Robjant and Fazel 2010 for a review on evidence for the effectiveness of NET).
4.2 Genetic Factors in the Etiology, Symptomatology, and Treatment of PTSD
As summarized above, traumatic load increases the risk to develop PTSD in a dose-dependent manner. Yet, individual risk factors are known to interact with traumatic load and influence the likelihood of subsequent PTSD development. From a biological perspective, it is hence crucial to look at genetic and epigenetic risk factors for PTSD.
4.2.1 Heritability of PTSD
Family studies examining biological relatives of patients diagnosed with PTSD first indicated genetic risk factors in disorder etiology. For example, children of PTSD patients from different contexts (Holocaust survivors, Cambodian refugees) were found to be at greater risk for PTSD following trauma exposure (Sack et al. 1995; Yehuda et al. 2001). Given that families share genetic and environmental influences (e.g., observational learning and parental distress), twin studies are needed to examine the heritability of PTSD more closely. Comparing PTSD in identical or monozygotic (MZ) twin pairs with those in fraternal or dizygotic (DZ) twin pairs results in heritability estimates of PTSD around 30–40 % (Stein et al. 2002; True et al. 1993), a figure which corresponds with estimates for anxiety disorders (Hettema et al. 2003). However, caution is needed, as identical twins may share a more similar exposure to stressful experiences in comparison to fraternal twins.
Whereas twin studies are useful to estimate the heritability of a disorder or trait, association studies are needed to identify which genes and, hence, physiological mechanisms are involved in the etiology of a certain disorder. Candidate gene association studies compare the genotype frequencies of polymorphic regions of certain candidate genes between affected individuals and healthy controls. Polymorphic regions of the genome are defined as regions that vary naturally between individuals. The most commonly studied polymorphisms are single nucleotide polymorphisms (SNPs; variation in a single base pair) and variable number of tandem repeat polymorphisms (VNTRs; the number of repeats and, hence, the length of a repetitive region of the genome differ). As a consequence, both polymorphisms can lead to functional differences in the gene-expression product.
Since PTSD requires exposure to an environmental stressor to manifest, the standard case-control design is not adequate for PTSD research, since controls might also carry genetic risk factors which never had an impact because the individual was not sufficiently exposed to traumatic stressors. Therefore, to understand genetic susceptibility of PTSD, it is crucial to carefully assess traumatic load and include it in the subsequent genetic analyses (Cornelis et al. 2010; Wilker and Kolassa 2013).
4.2.2 Genetics of the Fear Network
According to the fear network model detailed above (Elbert and Schauer 2002; Kolassa and Elbert 2007), strongly interconnected, highly accessible emotional-sensory fear memories, which are detached from the corresponding autobiographical context information, lead to intrusive PTSD symptoms (Wilker and Kolassa 2013).
Accordingly, fear memory formation, and fear conditioning in particular, has been widely used as a model for PTSD development. Fear conditioning is a prominent paradigm in PTSD research, since it can be easily studied in animals and provides a sound explanation on how previously neutral stimuli (e.g., a policeman) can become triggers of fear due to a traumatic experience. The hippocampus, the medial prefrontal cortex (mPFC), and the amygdala (together termed limbic-frontal neurocircuitry of fear) were identified as the main areas involved in the acquisition and regulation of conditioned fear in animal studies. The interplay of these areas during (traumatic) stress is influenced by the neuromodulatory actions of neurotransmitters, such as serotonin, dopamine, and norepinephrine, as well as hormones, such as glucocorticoids (for reviews see, e.g., Ressler and Nemeroff 2000; Rodrigues et al. 2009; Shin and Liberzon 2009). Therefore, these systems have been targeted by candidate gene association studies on PTSD.
Whereas studies only comparing the prevalence of genetic risk alleles in PTSD patients and controls have yielded conflicting results, research which accounts for the aforementioned building block effect and, hence, investigates gene × environment interactions has brought converging evidence for several genetic risk factors which will be summarized in the following.
4.2.2.1 Modulators of the Serotonergic System
The neurotransmitter serotonin is known to influence emotional learning and memory through its inhibitory action on the amygdala (cf. Meneses and Liy-Salmeron 2012; Ressler and Nemeroff 2000). The serotonin transporter is responsible for the active clearance of serotonin from the synaptic cleft. Among candidate genes encoding proteins involved in the serotonergic system, there is convergent evidence indicating an influence of the serotonin transporter gene on the risk to develop PTSD subsequent to traumatic stress. A length polymorphism within the promoter region of the serotonin transporter gene (termed serotonin transporter gene-linked polymorphic region; 5-HTTLPR) influences its activity: in contrast to the long (l) allele, the short (s) allele is associated with lower gene transcription and, hence, lower serotonin transporter activity, higher amygdala reactivity to emotional stimulation, and enhanced fear conditioning (Greenberg et al. 1999; Heils et al. 1996; Lonsdorf et al. 2009; Munafò et al. 2008). Studies investigating gene × environment interactions consistently show that the short variant enhances PTSD susceptibility subsequent to traumatic stress (Kilpatrick et al. 2007; Kolassa et al. 2010a; Mercer 2012; Pietrzak et al. 2013; Xie et al. 2009, 2012). To give an example, in a sample of Rwandan genocide survivors, carriers of the long variant of the serotonin transporter polymorphism showed the typical building block effect: the more traumatic events experienced, the higher the prevalence of PTSD. Yet, this cumulative effect was not seen in individuals carrying two copies of the short alleles, who already developed PTSD at relatively low traumatic load (Kolassa et al. 2010a; cf. Fig. 4.2).
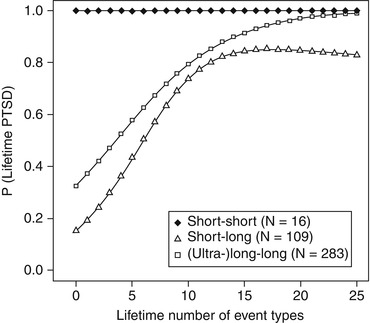
Fig. 4.2
Fitted values of probability for lifetime PTSD plotted against the number of traumatic event types for different genotypes (From Kolassa et al. (2010a), reprinted with permission)
Interestingly, the short allele was also associated with higher risk of PTSD symptom relapse 6 months after the completion of an 8-week trauma-focused cognitive behavior therapy (Bryant et al. 2010), suggesting more persistent fear memories in carriers of this risk genotype. Furthermore, PTSD patients who were homozygous for the long allele showed higher responsiveness to a pharmacological PTSD treatment with sertraline as well as a lower dropout rate (Mushtaq et al. 2012). Whereas there are only few studies investigating the influence of genetic polymorphisms on PTSD treatment success, this first evidence might indicate the need of personalized treatments depending on individual biological risk factors.
4.2.2.2 Modulators of the Dopaminergic System
Dopamine is another neuromodulator of the neurocircuitry of fear. More precisely, dopaminergic influx in the amygdala is important for the consolidation of fear memories (Guarraci et al. 2000). Dopaminergic activity is modulated by the enzyme catechol-O-methyltransferase (COMT), which degrades and thereby deactivates dopamine and other catecholamines. A SNP in the COMT gene results in the integration of the amino acid methionine (Met) instead of valine (Val) in the resulting protein (COMT Val158Met polymorphism). On the functional level, the Met allele is associated with lower COMT enzyme activity and, therefore, higher extracellular dopamine levels (Lachman et al. 1996), as well as impaired fear extinction learning (Lonsdorf et al. 2009). Similarly, the Met allele has been found to interact with traumatic load to predict higher risk for the development of PTSD (Boscarino et al. 2011; Kolassa et al. 2010c). Furthermore, there is initial evidence that this polymorphism might influence therapeutic treatment success. To date, only one study has investigated the influence of COMT genotype on psychotherapeutic outcome in a sample of panic disorder patients and found that Met/Met carriers benefited less from the exposure-based elements of the treatment (Lonsdorf et al. 2010). Since effective PTSD treatments also require exposure to the traumatic experiences, it would be interesting to investigate whether these findings translate to PTSD.
4.2.2.3 Modulators of the Biological Stress Responses
During a stressful or traumatic experience, the body’s alarm response is initiated by two major stress axes, the hypothalamus-pituitary-adrenal (HPA) axis and the locus coeruleus noradrenergic (LCNA) system. Their joint activity prepares the body for survival, e.g., by mobilizing glucose, increasing the heart rate and blood pressure, and enhancing muscle tension.
The HPA axis activation consists of three steps: initially, corticotropin-releasing hormone is released from the hypothalamus. This stimulates the pituitary gland to secrete adrenocorticotropic hormone, which leads to the release of cortisol from the adrenal glands. Whereas this stress response is initially adaptive, a prolonged HPA axis activity can lead to adverse health consequences. In order to avoid chronic HPA axis activation, the binding of cortisol to glucocorticoid receptors exerts negative feedback on the release of further corticotropin-releasing hormone and adrenocorticotropic hormone from the hypothalamus and pituitary, respectively. Elevated cortisol levels have been further found to increase the memorization of emotional experiences, rendering the glucocorticoid system an interesting target for candidate gene association studies on PTSD (see, e.g., Wolf 2009 for a review). Yet, studies investigating glucocorticoid receptor polymorphisms have yielded inconsistent results (Bachmann et al. 2005; Hauer et al. 2011). Another interesting candidate in the glucocorticoid system is the gene encoding co-chaperone FK506-binding protein 51 (FKBP5), which regulates cortisol-binding affinity of the glucocorticoid receptor. More precisely, when FKBP5 binds to co-chaperone FK506, the resulting receptor complex decreases the binding capacity for cortisol (Binder 2009), resulting in reduced negative feedback and, hence, a prolonged stress reaction. So far, research consistently points towards an interaction of FKBP5 risk alleles and adult or childhood trauma exposure in the risk to develop PTSD (Binder et al. 2008; Boscarino et al. 2011, 2012; Xie et al. 2010).
Stress or emotional arousal leads to the release of noradrenaline from the basolateral amygdala, and this noradrenergic neurotransmission is required to form memories of emotional experiences (e.g., McGaugh and Roozendaal 2002). PTSD is associated with both central and peripheral noradrenergic hyperactivity (Heim and Nemeroff 2009), yet few studies investigated genes involved in the noradrenergic system. One interesting exception is the work of de Quervain and coworkers (2007), who investigated a deletion variant in the gene ADRA2B, coding the alpha-2B-adrenergic receptor, which was associated with enhanced memory for emotionally arousing material in healthy volunteers and more pronounced intrusive memories in survivors of the Rwandan genocide (de Quervain et al. 2007).
In sum, reliable genetic associations were found with genes involved in the development of pathological memories. Understanding genetic risk factors relevant to the formation of intense fear memories is especially important, since those biological systems present a promising target for modifications through future treatment opportunities, especially on a pharmacological level.
Yet, despite increasing efforts and technological advances, only a small proportion of the estimated PTSD heritability can be explained by the identified genetic risk factors. Explanations for this “missing heritability” (Manolio et al. 2009) include the presence of complex multigene interactions and interactions between the individual environmental exposure with the genetic makeup of a person. Furthermore, research in the last decades has elucidated that environmental exposure can shape gene expression through epigenetic modification (Zhang and Meaney 2010). The next section will hence illuminate epigenetic mechanisms, which are essential to obtain a full understanding of the interaction of nature and nurture in PTSD etiology.
4.3 Epigenetic Alterations Associated with PTSD
The term epigenetics originates from the Greek syllable epi, meaning upon, and genetics. The epigenome can be viewed as a “second layer of information” (Zhang and Meaney 2010, p. 447) which consists of chemical modifications altering the accessibility of the DNA, without changing the sequence of nucleotides. More precisely, gene expression depends upon the binding of transcription factors to the promoter region of a gene, and different epigenetic mechanisms dynamically allow or prevent transcription factor binding. These mechanisms are crucial to cell differentiation: since the genome of mostly all cells in a given human individual is the same, the epigenetic profile allows the specialization regarding the cellular function (e.g., heart cell vs. neuron) by determining which genetic sequences are expressed as proteins. Furthermore, and of particular relevance in the context of PTSD, epigenetic alterations allow for the dynamic adaption of gene expression to challenging environmental demands (Zhang and Meaney 2010). In the last years, evidence suggesting that traumatic stress can influence the individual epigenetic profile has accumulated (Malan-Müller et al. 2013). At first, it was supposed that particularly early developmental adversity leads to alteration in the epigenome, which can be associated with higher stress vulnerability later in life. Thereafter, however, it was acknowledged that epigenetic modifications occur during the entire lifespan and are crucial to learning and memory (Zhang and Meaney 2010). Hence, on the one hand, early developmental stress can lead to stable epigenetic alterations which constitute a risk factor for later PTSD development. On the other hand, adult traumatic stress might also lead to epigenetic modifications which could in turn perpetuate the psychological symptoms and biological alterations associated with PTSD.
The three epigenetic mechanisms which may lead to an alteration of the transcriptional accessibility of genes without changing their structure are RNA-associated silencing, histone modification, and most prominently DNA methylation (Egger et al. 2004). The modifications can enhance or decrease gene expression and even “silence” the gene entirely (Sutherland and Costa 2003).
First evidence for the influence of early experiences on the epigenetic profile stems from animal studies which show that low versus high maternal care of rat mothers leads to differential DNA methylation in their offspring (Weaver et al. 2004). More precisely, low maternal care was associated with enhanced methylation of the glucocorticoid receptor gene (GCCR) promoter in the hippocampus and, hence, reduced GCCR expression. As indicated earlier, the GCCR is involved in negative regulatory feedback of the hypothalamic-pituitary-adrenal (HPA) axis. Increased GCCR promoter methylation can therefore reduce GCCR sensitivity, which might lead to a prolonged HPA activation under stress. Returning to the study by Weaver et al. (2004), these epigenetic changes, which may persist into adulthood, could be prevented by placing the rodents with rat mothers providing them with high maternal care – a finding that is of utmost importance for psychotherapy in the field of psychotrauma.
McGowan et al. (2009) showed comparable findings in humans: they found enhanced methylation in a neuron-specific glucocorticoid receptor gene (NR3C1, the human homolog of the site investigated in rats in the Weaver study) promoter in the hippocampi of suicide victims who had experienced childhood abuse, compared to suicide victims with no documented history of childhood abuse. The study implies a common effect of early stress experiences on the epigenetic regulation of hippocampal GCCR expression (McGowan et al. 2009). This was confirmed by a recent study (Mehta et al. 2013) comparing DNA methylation and gene expression between a sample of PTSD patients with childhood and adult trauma, a PTSD sample without childhood trauma, and a trauma-exposed control group without PTSD. The authors found distinct gene expression patterns in the two PTSD groups, indicating differential biological pathways involved in PTSD etiology depending on childhood trauma history. Furthermore, only in the childhood trauma group, differences in gene expression overall matched with epigenetic modifications of the corresponding gene loci, suggesting that childhood trauma leads to enduring epigenetic alterations which still influence gene expression in adulthood.
Likewise, Radtke et al. (2011) showed for the first time that prenatal stress – in particular intimate partner violence – experienced by mothers during pregnancy altered the methylation status of the GCCR of their offspring (assessed at age 10–19). Hence, this study provides a possible link between prenatal stress and psychopathology later in life (Radtke et al. 2011).
Indeed, recent studies provide initial evidence that epigenetic modifications influence the probability of PTSD development. For instance, Koenen and colleagues showed in a primarily African American sample from the Detroit Neighborhood Health Study that methylation level at the serotonin transporter promoter polymorphisms (5-HTTLPR) and traumatic load interacted to predict PTSD vulnerability (Koenen et al. 2011). Chang and colleagues investigated the dopamine transporter gene, another genetic locus previously found to be implicated in PTSD vulnerability (Drury et al. 2009; Segman et al. 2002; Valente et al. 2011; but see Bailey et al. 2010). They found the highest PTSD probability in carriers of the high-risk genotype, who also present with higher methylation levels at this locus (Chang et al. 2012). Moreover, it was recently suggested that the combination of an FKBP5 risk genotype and early adversity leads to demethylation at the FKBP5 locus, which may be associated with higher FKBP5 responsiveness and an augmented stress response later in life (Klengel et al. 2012). These studies provide first evidence that traumatic experiences, especially if they occur during early developmental windows, can shape the epigenetic profile, which in turn influences the vulnerability for PTSD in the case of subsequent trauma exposure. Furthermore, the work of Klengel and coworkers indicates that epigenetic alterations may be influenced by genotype. Consequently, we are confronted with a complex interaction of genetics, epigenetics, and early and late trauma exposure in the prediction of PTSD.
4.4 Structural and Functional Alterations in the Brain of Trauma Survivors with PTSD
Structural changes associated with PTSD such as hippocampal atrophy could be the cause for functional impairments and dysfunctions associated with PTSD (Sherin and Nemeroff 2011) but were also discussed as risk factors (Gilbertson et al. 2002).
Using brain-imaging methods, PTSD has been implicated with structural changes in the medial prefrontal cortex (mPFC), the amygdala, and the hippocampus (see Liberzon and Sripada 2008 for a review). The mPFC performs inhibitory control over stress responses through its connection to the amygdala, and it plays a major role in fear extinction (Heim and Nemeroff 2009). According to the neurocircuitry model of PTSD, regions of the ventral mPFC, which are supposed to inhibit the amygdala, are dysfunctional in patients suffering from the disorder. At the same time as the amygdala responses are amplified, the hippocampus shows deficits in explicit learning and memory and, therefore, fails to identify safe environments (Rauch et al. 2006). The hippocampus (Fig. 4.3) is especially important in the development of PTSD; it is crucial for controlling stress responses as well as declarative memory, and it plays a major role in contextual aspects of fear conditioning (Heim and Nemeroff 2009). Since this area of the brain is known for its high plasticity, studies investigating structural changes in the brain due to PTSD have mainly focused on volume changes in the hippocampus (Hughes and Shin 2011). Animal studies show that pathological stress can lead to hippocampal size reduction, possibly caused by adverse effects of stress hormones (Sapolsky et al. 1990). Several meta-analyses describe smaller hippocampal volumes in trauma-exposed individuals with and without PTSD, when compared to a non-exposed control group (Smith 2005; Woon et al. 2010), while others find hippocampal volume reduction only in PTSD patients but not in trauma-exposed and non-exposed control groups (Karl et al. 2006; Kitayama et al. 2005).
Fig. 4.3
Morphological alterations of the hippocampus have been repeatedly found in PTSD and might represent a risk factor or a consequence of the disorder
Gilbertson and colleagues (2002) conducted a study with twins; they compared war veterans with combat exposure to their twins without combat exposure. In accordance with previous studies, they also found smaller hippocampi in PTSD patients. However, the co-twins of the PTSD patients who neither suffered from PTSD nor had been exposed to combat also had smaller hippocampi compared to the non-PTSD control group (Gilbertson et al. 2002). This suggests smaller hippocampal volume to be a risk factor for PTSD rather than a consequence of the disorder. In a further study, configural processing performance was found to be significantly lower in PTSD patients in comparison to trauma-exposed controls. Nevertheless, the co-twins of the PTSD patients showed the same impairments which were related to hippocampal volume, even though they did not suffer from PTSD, nor were they trauma-exposed as adults (Gilbertson et al. 2002). However, a recent study of Teicher et al. (2012), which presents evidence that childhood adversity massively affects hippocampal development, provides an alternative explanation for the results of the Gilbertson work group. It is possible that those twins exposed to adversity during vulnerable childhood periods may have both a reduced hippocampal volume and a greater risk for developing PTSD when exposed to combat. Possibly such altered development of hippocampal organization may reduce contextualizing representations of stressful events and generalize fear. While this brain organization may be adaptive in dangerous environments, it also includes the risk of extending the fear network and eventually the maladaptive formation of PTSD.
Finally, another study found reduced hippocampal volume in current PTSD patients compared to a control group with remitted PTSD. Again, two alternative explanations could account for this finding: on the one hand, lower hippocampus volume could be a risk factor for a chronic pathology of PTSD. On the other hand, smaller hippocampus volume could be a consequence of PTSD, which might be reversible if the disorder remits (Apfel et al. 2011).
To conclude, the current state of research does not clearly support the notion that reduced hippocampus volume is a consequence of traumatic stress or a (genetic) risk factor for PTSD onset. Yet, an association of PTSD with reduced hippocampal volume is evident and corresponds well with the assumption of unreliable autobiographical context information of the fear network model.
Since the amygdala plays a central role in the neurocircuitry of fear in general, and the acquisition and expression of conditioned fear reactions in particular, several studies looked at structural alterations of the amygdala in survivors suffering from PTSD. Yet, in contrast to research on hippocampus volume, results are far from consistent. For instance, a meta-analysis from 2009 which summarized nine studies found no differences in left and right amygdala volume if PTSD patients were compared to trauma-exposed or unexposed control groups (Woon and Hedges 2009). More recent studies continue to provide inconsistent findings. Kuo and colleagues (2012) found enhanced total amygdala volumes in combat veterans with PTSD compared to trauma-exposed veterans without PTSD. Yet, another study also investigating trauma-exposed military veterans found reduced left and right amygdala volume in PTSD (Morey et al. 2012).
Finally, PTSD-associated structural alterations of the PFC, which plays a central role in fear inhibition and extinction learning through its inhibitory influence on the amygdala, have been investigated. A recent meta-analysis summarized the results of nine studies using voxel-based morphometry to assess gray matter volume in PTSD patients opposed to trauma-exposed controls and found significantly reduced volumes in several regions implicated in the neurocircuitry of fear, including the left hippocampus and the ventromedial PFC (Kühn and Gallinat 2013).
In addition to the identified structural alterations in the neurocircuitry of fear, it is also of interest to understand if the functioning or the interplay of these structures is altered in PTSD patients. A consistent body of research indicates that amygdala reactions to threatening cues are accelerated in PTSD. The common design to investigate amygdala reactivity is to compare brain activity while presenting neutral versus emotional stimuli. For instance, Brohawn and coworkers (2010) found significantly elevated amygdala reactivity in response to negative pictures in PTSD patients compared to trauma-exposed controls. These results were confirmed in numerous studies, revealing higher amygdala activity in response to fearful or aversive stimuli in PTSD patients versus controls (see Hayes et al. 2012 for a recent meta-analysis), as well as a positive correlation between current PTSD symptom severity and amygdala reactivity to these stimuli (Armony et al. 2005; Dickie et al. 2008, 2011). In addition, a recent study found enhanced spontaneous amygdala activity in PTSD, indicating that an increase in amygdala activity in PTSD is not only present during specific experimental provocations but can be a general condition in PTSD (Yan et al. 2013). Heightened amygdala activity is associated with decreased mPFC activity in response to aversive stimuli (Hayes et al. 2012). This is supplemented by recent evidence indicating that trauma survivors with PTSD show reduced functional connectivity between the amygdala and the mPFC in response to fearful stimuli, a finding that can further explain the failure of the mPFC to inhibit exaggerated amygdala responses in PTSD (Stevens et al. 2013).
Somewhat in contrast to the general finding of prefrontal hypoactivity in PTSD, Adenauer and colleagues found enhanced early activation in the right prefrontal cortex in response to aversive pictures in PTSD patients, compared to trauma-exposed individuals not suffering from PTSD and unexposed control subjects (Adenauer et al. 2010). Such early activity might be missed in functional magnetic resonance imaging studies, which have a lower time resolution than the magnetoencephalography (MEG) paradigm implemented in the Adenauer study. This early prefrontal activity might represent an acquired hypersensitivity to threatening stimuli in a traumatized brain, which constitutes one form of expression of a strong fear network that is prepared to rapidly detect danger (Rockstroh and Elbert 2010). This early adaptive activation is most likely followed by subsequent deactivation of prefrontal regions, a vigilance-avoidance pattern that was also described for other anxiety disorders (Adenauer et al. 2010).
4.5 The Effects of Psychological Trauma on Physical Health: Identifying Potential Molecular Modulators
PTSD has been associated with poor self-reported health and increased healthcare utilization (Schnurr and Jankowski 1999) but also unfavorable lifestyle factors such as physical inactivity and smoking (Zen et al. 2012). Psychological stress, particularly if it takes extreme (traumatic) forms, has been shown to enhance the risk for cardiovascular, cerebrovascular, respiratory, gastrointestinal, musculoskeletal, inflammatory, and autoimmune diseases, as well as other age-related diseases including even cancer (Boscarino 2004; Felitti et al. 1998; Fuller-Thomson and Brennenstuhl 2009; Glaesmer et al. 2011; Schnurr and Jankowski 1999). For more details, see Chap. 5.
Although traumatic experiences and PTSD have often been related to adverse health outcomes, the precise molecular mechanisms underlying this relationship warrant further investigation. The ongoing stress an individual is exposed to through adverse environments (traumatic stressors) seems to cause the immune system to age prematurely and to alter various molecular pathways. Sommershof et al. (2009) showed a relative reduction in naïve cytotoxic and regulatory T cells in the lymphocytes of PTSD patients as well as a relative increase in memory T cells due to more past infections and a higher wear and tear of the immune system. The reduction in naïve cytotoxic T cells might lead to a compromised immune response, which could then contribute to the enhanced susceptibility for infections. A similar effect is observed in elderly people, where the natural reduction of naïve T lymphocytes leads to a greater risk for diseases associated with aging (Shen et al. 1999). In addition, the reduction of regulatory T cells might put PTSD patients at risk for inflammatory and autoimmune diseases. Indeed, there is a high prevalence of autoimmune and inflammatory diseases in PTSD patients (Boscarino 2004). More interestingly, we observed in this study (Sommershof et al. 2009) and in an extension study (Morath et al. 2014a) that naïve cytotoxic T cells and regulatory T cells were also reduced in trauma-exposed individuals without a diagnosis of PTSD; this group took an intermediate position between PTSD patients and controls. As traumatized individuals with PTSD usually experienced more traumatic events than traumatized individuals without PTSD, this suggests a cumulative effect of traumatic stress on T-cell distribution independent of the diagnosis of PTSD (Sommershof et al. 2009).
Related posts:
 EMDR Therapy for Trauma-Related Disorders
Complicated Grief Treatment (CGT) for Prolonged Grief Disorder
Psychological and Social Theories of PTSD
EMDR Therapy for Trauma-Related Disorders
Complicated Grief Treatment (CGT) for Prolonged Grief Disorder
Psychological and Social Theories of PTSD
 The Complexity of Chronic Pain in Traumatized People: Diagnostic and Therapeutic Challenges
The Complexity of Chronic Pain in Traumatized People: Diagnostic and Therapeutic Challenges
 Narrative Exposure Therapy (NET): Reorganizing Memories of Traumatic Stress, Fear, and Violence
What Works for Whom?
Narrative Exposure Therapy (NET): Reorganizing Memories of Traumatic Stress, Fear, and Violence
What Works for Whom?
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


