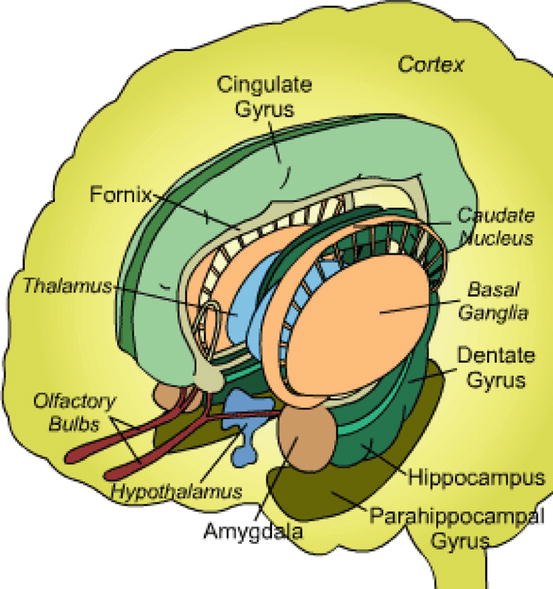
Fig. 14.2
Anatomy of the limbic system: from the Huntington’s Outreach Program for Education at Stanford, www.stanford.edu
A range of work supports an amygdalocentric model. Several studies demonstrate increased amygdalar activity in response to emotional stimuli in PTSD (Rauch et al. 1996, 1997; Shin et al. 1997a; Liberzon et al. 1999; Pissiota et al. 2002; Vermetten et al. 2007). A symptom provocation study using [oxygen-15]-carbon dioxide (15O-CO2) PET found that symptom severity was positively related to rCBF changes in the right amygdala (Shin et al. 2004a). Similarly, in an [oxygen-15]-water (15O-water) PET study, during habituation, acquisition and extinction conditions, PTSD patients were found to have increased activation in the left amygdala with fear acquisition relative to the control group (Bremner et al. 2005). However, findings of amygdala hyperactivity in PTSD are not found in all studies (Britton et al. 2005; Phan et al. 2006; Molina et al. 2010).
Recent nuclear neuroimaging studies have also identified the insula, a region closely related to the amygdala, as hyperactive in PTSD. The insula is heavily interconnected with the amygdala, hypothalamus and periaqueductal gray matter (Paxinos 2003) and regulates the autonomic nervous system (Oppenheimer et al. 1992) as well as being involved in the processing of negative emotions (Phan et al. 2002). Hyperactivity of this region during emotional processing (Osuch et al. 2001; Lindauer et al. 2004; Nardo et al. 2011) most likely represents activation of a network (incorporating amygdala) responsible for generating fear responses to symptom-provoking stimuli (Etkin and Wager 2007). A rest study found a positive correlation between insular activity (as measured by 15O-water) and the intensity of flashbacks with symptom provocation (Osuch et al. 2001). Increased insular activity in PTSD was noted in meta-analyses of PET and fMRI data (Etkin and Wager 2007; Hayes et al. 2012). A positive correlation has also been reported between insular activity and ACTH response in a symptom provocation study in PTSD (Liberzon et al. 2007a) [see later section]. Not all studies support insular hyperactivity in PTSD however (Shin et al. 1997a, b, 1999; Lindauer et al. 2004; Fernandez et al. 2001; Molina et al. 2010).
There is growing work on the precise role of the vmPFC in PTSD. In addition to the evidence that insufficient influence by vmPFC underlies deficits in extinction as well as the capacity to suppress attention and response to trauma-related stimuli in PTSD (Rauch et al. 2006; Bremner et al. 1999a, b, 2005; Shin et al. 1999; Lindauer et al. 2004), a number of studies reveal the inverse relationship between vmPFC and amygdala activity in these patients. In a symptom provocation study using 15O-CO2 PET, rCBF changes in the medial frontal gyrus were inversely correlated with rCBF changes in the left amygdala and the right amygdala/periamygdaloid cortex. Symptom severity was also negatively related to reductions in rCBF in the medial frontal gyrus (Shin et al. 2004a). The negative correlation between amygdalar activity and vmPFC activity in PTSD was also reported in a recent meta-analysis of PET and fMRI studies (Hayes et al. 2012). Such an inverse relationship between vmPFC and amygdala has not been replicated by all studies (Gilboa et al. 2004; Gold et al. 2011).
The role of the anterior cortex, including the vmPFC, in PTSD is probably complex and may be better understood by understanding it as a number of substructures with interrelated roles. The vmPFC includes rostral anterior cingulate cortex (rACC), medial prefrontal cortex (mPFC), subcallosal cortex and orbitofrontal cortex. The anterior cingulate cortex (ACC) and vmPFC are overlapping regions, sharing the rACC. The gating function ascribed to the vmPFC in modulating conditioned fear responses (Rauch et al. 1998) has been ascribed to the ACC (Hamner et al. 1999). This latter model, largely based on animal research (e.g. Sparenborg and Gabriel 1992), proposed an amygdala-locus coeruleus-anterior cingulate circuit whereby efferent noradrenergic projections from the locus coeruleus may dampen the anterior cingulate function, thereby allowing external and internal stimuli to produce the exaggerated emotional and behavioural responses characteristic of PTSD (Fig. 14.3).
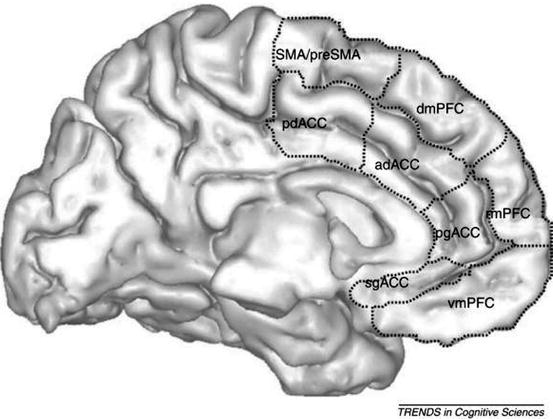
Fig. 14.3
Subdivisions of the mPFC and ACC. Abbreviations: sg subgenual, pg pregenual (rostral), vm ventromedial, rm rostromedial, dm dorsomedial, ad anterior dorsal, pd posterior dorsal (From Etkin et al. (2011))
Examining the activation of the ACC as a whole is probably too simplistic, since there is evidence for functional specialisation of subregions of the ACC. The rostral anterior cingulate cortex (rACC) is involved in the assessment of emotional information as well as the regulation of emotional responses (Mohanty et al. 2007). There is evidence that the rACC has a gating function as proposed by Hamner et al. (1999) and this is consistent with the amygdalocentric model (Rauch et al. 1998). The majority of nuclear neuroimaging studies have reported reduced rCBF (on 15O-water PET) to rACC in PTSD patients during symptom provocation (Bremner et al. 1999a, b; Britton et al. 2005). A study that used 15O-water PET with a cognitive activation paradigm also reported diminished rCBF in rACC in PTSD compared to trauma-exposed controls (Bremner et al. 2004). Some symptom provocation studies have however reported either decreased (Shin et al. 1997a; Liberzon et al. 2007a) or equal activation (Liberzon et al. 1999) in rACC in PTSD patients compared to control groups.
The dorsal anterior cingulate cortex (dACC) appears to be involved in cognitive processing including conflict monitoring, response selection, error detection (Bush et al. 2000; Carter et al. 1999), aversive conditioning (Milad et al. 2007; Büchel et al. 1998) and the anticipation and perception of pain (Chua et al. 1999; Derbyshire 2000). Subregional analysis has yielded the dACC as an additional node of potential importance in PTSD. Most nuclear neuroimaging research supports the hyperactivation of dACC in PTSD. In a study using [technetium-99m] ethyl cysteinate dimer (99mTc ECD) SPECT under rest conditions, it was reported that PTSD patients demonstrated increased rCBF in dACC compared to healthy controls (Chung et al. 2006). A study that used identical twins as control subjects performed 2-deoxy-2-[fluorine-18] fluoro-d-glucose (18F-FDG) PET to compare PTSD patients, trauma-exposed controls and healthy controls under rest conditions and reported that in the PTSD group, there was relatively increased activity in dACC (Shin et al. 2009). Mid- and dorsal anterior cingulate cortex was one of the regions most consistently hyperactivated in PTSD patients in a recent meta-analysis (Hayes et al. 2012), which identified dmPFC (including mid/dorsal ACC) as one of the several key salience network regions. Some symptom provocation studies have however reported deactivation (Shin et al. 1999; Gold et al. 2011) or equal activation (Phan et al. 2006) in dACC in PTSD patients compared to control groups.
Because PTSD is associated with memory problems, abnormal hippocampal function has frequently been considered in neuroimaging studies. This structure ordinarily provides information about the context of a situation through its role in explicit memory, and dysfunction of the hippocampus may help explain memory problems in the disorder as well as deficits in identifying safe contexts (Bremner et al. 2005). Debate exists regarding possible mechanisms of purported hippocampal dysfunction, which include chronic stress of PTSD being toxic to the hippocampus, and variation in amygdalar influence on hippocampal function (Elzinga and Bremner 2002; Layton and Krikorian 2002). The nuclear neuroimaging literature regarding hippocampus in PTSD is controversial, reporting activations (Chung et al. 2006; Britton et al. 2005; Shin et al. 2004b; Yehuda et al. 2009), deactivations (Molina et al. 2010; Kim et al. 2012; Bremner et al. 1999a, 2003a, b) or no change (Rauch 1996) in hippocampal activity. A PTSD treatment study using 99mTc-ECD SPECT found hippocampal activation after psychotherapy (Peres et al. 2007). Meta-analysis of PET and fMRI studies reveals no change (Hayes et al. 2012). The variation in reported results may in part be related to chronicity of the disorder, in which hippocampal deactivation is a feature of chronic PTSD (Bremner et al. 2003a).
Limited nuclear neuroimaging research has specifically examined the relationship between hippocampal dysfunction and memory deficiencies in PTSD. A cognitive activation study to test verbal declarative memory performed using 15O-water PET to scan patients with PTSD, trauma-exposed controls and healthy controls reported no significant differences in memory performance between the groups, but it was found that the PTSD group demonstrated failure of activation of the left hippocampus during encoding (Bremner et al. 2003a). A study using 15O-water PET to scan PTSD patients and healthy controls using a visuoverbal target detection task involving continuous updating (variable target condition) or no updating (fixed target condition) to examine verbal working memory in PTSD reported no hippocampal findings (Clark et al. 2003).
Located in the left inferior frontal cortex (BA 44, 45), Broca’s area is the structure believed to be responsible for applying semantic representations to personal experience which allow communication or description of this experience (Hull 2002). Various groups have implicated deactivation of Broca’s area to explain why PTSD patients struggle to cognitively restructure their traumatic experience (Rauch et al. 1996; Shin et al. 1997a, b, 1999; Osuch et al. 2001; Bremner et al. 2003a; Lindauer et al. 2004). Treatment studies using both non-pharmacologic (Lansing et al. 2005; Peres et al. 2007) and pharmacologic therapy (Fernandez et al. 2001) report activation of Broca’s area in PTSD patients after therapy.
14.3 Neurochemistry of PTSD
There is a growing understanding of the neurochemistry of the stress response as well as of PTSD (Chrousos 2009). The hypothalamic-pituitary axis (HPA), the sympathetic adrenomedullary system and a range of other neurotransmitters play a key role. Receptor-radioligand imaging is one of the more promising means of assessing these systems although it is not without its limitations: altered binding of a radioligand may be explained by several different mechanisms which may be difficult to distinguish (reduced radioligand delivery, altered receptor affinity, altered number of receptors, altered concentrations of endogenous competing molecules or altered removal of radioligand from the receptor site). Currently, there are limited molecular imaging data available, but they are already assisting in understanding PTSD from a neurochemical perspective, and this work is likely to be key to improving our understanding of the neurobiology of PTSD in the future. In this section we will cover existing molecular imaging work on neurochemical/neuroreceptor systems implicated in PTSD.
14.3.1 Serotonergic System
Animal research has implicated the serotonin system in anxiety – specifically in the modulation of anxiety and fear. Inescapably shocked animals develop decreased levels of CNS serotonin (Valzelli 1982). In animal studies, low serotonin is related to an inability to modulate arousal (Gerson and Baldessarini 1980; Dupue and Spoont 1989) – an observation that may be relevant to the phenomenology of PTSD in humans (van Der Kolk 1994). Evidence for a role of the serotonergic system in PTSD is also to be found in the efficacy of selective serotonin reuptake inhibitors (SSRIs) in treating the symptoms of the disorder (Albucher and Liberzon 2002).
A study that used [technetium-99m]-d, l-hexamethyl-propyleneamine oxime (99mTc-HMPAO) SPECT to scan 11 patients with PTSD pre and post 8 weeks of treatment with citalopram (a SSRI) found that treatment with citalopram resulted in significant deactivation in the left medial temporal cortex (incorporating hippocampus and amygdala) irrespective of clinical response (Seedat et al. 2004). A significant correlation was found between PTSD symptom improvement and activation in the left paracingulate region (mPFC). These data are consistent with a model in which treatment of PTSD with a SSRI may extinguish learned fear responses by restoring the normal mPFC modulation of amygdala activity. The findings in the mPFC are supported by a study which used 15O-water PET to measure rCBF in 13 PTSD patients before and after 12 weeks of treatment with paroxetine (SSRI) or placebo. Patients were scanned with neutral and trauma-related scripts at each time point. While both placebo and paroxetine treatment was associated with a significant increase in rCBF to the ACC in response to traumatic scripts (as well as improvement in symptoms), paroxetine treatment was associated with an additional increase in function of the OFC (a component of vmPFC) (Fani et al. 2011).
Mouse knockout models have demonstrated the role of serotonin type 1A (5-HT1A) receptors in the regulation of anxiety responses (Parks et al. 1998; Ramboz et al. 1998). There is also evidence that the efficacy of SSRIs in treating anxiety is at least in part attributable of their binding to this inhibitory receptor (Varnas et al. 2004). This is growing evidence for the role of this receptor in anxiety (Akimova et al. 2009). Despite this evidence, a study using [fluorine-18]-trans-4-fluoro-N-2-[4-(2-methoxyphenyl) piperazin-1-yl]ethyl-N-(2-pyridyl) cyclohexanecarboxamide (18F-FCWAY) to compare 5-HT1A binding in 12 patients with PTSD and 11 healthy controls during rest conditions found no differences in binding between the groups in a priori defined areas of interest (midbrain raphe, anterior temporopolar cortex, posterior cingulate, anterior insula, anterior cingulate and medial temporal cortex (hippocampus plus amygdala) (Bonne et al. 2005). Their findings suggest that other serotonin receptor deficits or alternative alterations in serotonergic function need to be sought in PTSD.
A study of serotonin type 1B receptor (5-HT1B) expression has also been conducted by using [carbon-11]-P943 (11C-P943) to scan 49 patients with PTSD, 20 trauma-exposed controls and 27 healthy controls (Murrough et al. 2011a). Again, no significant differences were found between the PTSD group and the trauma-exposed controls, although marked reductions in ligand binding in the caudate, the amygdala and the ACC were seen in PTSD and trauma-exposed individuals compared to healthy controls – a finding which may be related to trauma exposure rather than PTSD per se.
Animal research focusing on the serotonin transporter (5-HTT) has demonstrated altered morphology of basolateral amygdala in 5-HTT knockout mice (Wellman et al. 2007; Hariri and Holmes 2006), whereas overexpression of the human 5-HTT gene in transgenic mice resulted in a low-anxiety phenotype (Jennings et al. 2006). Genetic studies in humans have demonstrated that the short allele of the common repeat polymorphism in the promoter region of the gene coding for the serotonin transporter (5-HTTLPR) increases vulnerability of developing PTSD (Lee et al. 2005; Kilpatrick et al. 2007; Xie et al. 2009; Kolassa et al. 2010). A study examining serotonin transporter binding using [carbon-11]-2-[2-(dimethylaminomethyl)phenylthio]-5-fluoromethyl-phenylamine (11C-AFM) in 15 patients with PTSD and 15 healthy controls found that ligand binding in the left amygdala was reduced in PTSD compared to the controls. Furthermore, amygdala ligand binding was inversely correlated with both anxiety and depression scores. No between-group differences for any other brain regions were detected (Murrough et al. 2011b). The group’s findings support the role of the amygdala in PTSD and are consistent with evidence from animal research that 5-HT signalling within the amygdala regulates normal fear and threat responsiveness (Wellman et al. 2007; Muller et al. 2011; Zanoveli et al. 2009). It may be that altered 5-HTT function influences amygdala activity to enhance the acquisition of conditioned fear and/or decrease fear extinction and that this mediates a vulnerability to PTSD (Murrough et al. 2011b).
14.3.2 Noradrenergic System
Preclinical evidence for the role of the noradrenergic system in stress is well established (Chrousos and Gold 1992; Charney et al. 1993). Noradrenergic cell bodies in the brain are predominantly concentrated in the locus coeruleus from which there are projections to cortical and subcortical structures (e.g. Cedarbaum and Aghajanian 1978; Ashton-Jones and Bloom 1981). Noradrenergic models of stress in animals have demonstrated evidence of locus coeruleus hyperactivity and resultant increase in release of noradrenaline in multiple brain regions (Redmond 1979; Tanaka et al. 1982). There has long been evidence to suggest that PTSD patients have increased sympathetic nervous system activity (see, e.g. Kosten et al. 1987).
No nuclear neuroimaging studies have thus far examined adrenergic receptors in PTSD directly, though the development of novel radioligands may allow this in the future (e.g. Van der Mey et al. 2006; Logan et al. 2007). Indirect evidence supporting the role of the adrenergic system in PTSD is to be found in a study in which ten patients with PTSD and ten controls underwent scans using 18F-FDG after yohimbine (an α2-antagonist) and after placebo administration. Of the ten PTSD patients, six had a panic attack with yohimbine administration and three had a flashback. None of the controls experienced symptoms. Significant differences in the brain metabolic response to yohimbine were seen between PTSD and controls in orbitofrontal cortex, temporal cortex and prefrontal cortex. Metabolism tended to decrease in patients with PTSD and increase in healthy subjects following administration of yohimbine (Bremner et al. 1997). These findings suggest that in PTSD either there is increased noradrenaline in central brain structures, which affects metabolism (and presumably symptoms), or there are alterations in specific noradrenergic receptors.
14.3.3 Gamma-Aminobutyric Acid (GABA) System
The efficacy of benzodiazepines in the treatment of anxiety disorders has led to interest in their underlying mechanisms of action. It has been known for decades (from animal research) that the GABAA receptor is the main target for the central actions of benzodiazepines (Costa et al. 1975; Haefely et al. 1975). Animal studies on benzodiazepine binding in conditions of inescapable stress have demonstrated reductions in the frontal cortex (e.g. Lippa et al. 1978; Weizman et al. 1989) and cerebral cortex (e.g. Medina et al. 1983), as well as (less consistently) in the hippocampus (e.g. Medina et al. 1983 vs. Weizman et al. 1989).
There is evidence that in PTSD there is reduced benzodiazepine binding in several brain regions. In a study using [iodine-123] iomazenil (123I-iomazenil) SPECT to study benzodiazepine receptor density in 13 PTSD patients and 13 healthy controls, it was reported that benzodiazepine receptor binding was 41 % lower in PTSD (compared to controls) in the dorsolateral prefrontal cortex. No other differences were detected between the two groups. A positive correlation was also found between symptom severity in the PTSD group and decreased benzodiazepine binding in the dorsolateral prefrontal cortex (Bremner et al. 2000). Another study that used [carbon-11] flumazenil (11C-flumazenil) dynamic PET to study benzodiazepine binding in nine patients with PTSD and seven trauma-exposed controls reported that PTSD subjects showed decreased ligand binding potential throughout the cortex, caudate nuclei, hippocampus, thalamus, left amygdala and left striatum in comparison to the control group (Geuze et al. 2008). The data may represent a difference in subunit composition of GABAA-benzodiazepine receptors in PTSD, a lower expression of GABAA receptor in PTSD or a disease or trauma-induced modulation or downregulation of the of GABAA-receptor complex (Geuze et al. 2008). Failure to demonstrate a global reduction in benzodiazepine-binding receptors by the former group may be due to differences in imaging modality. Nevertheless, not all data are consistent; a study that used 123I-iomazenil to image benzodiazepine receptor density in 19 PTSD patients and 19 healthy controls found no significant differences in ligand binding in any brain regions, even after partial volume correction (Fujita et al. 2004).
14.3.4 Opioid System
In animal studies, the endogenous opioid system has been implicated in the modulation of fear (Good and Westbrook 1995) as well as anxiolysis (Kang et al. 2000). Abnormalities in this system in PTSD have been proposed by several groups (Glover 1993; Pitman et al. 1990). Several studies have shown that the μ-opioid system has an inhibitory role during negative emotional states. Trauma exposure or recall may result in an exaggerated release of endogenous opioids with subsequent downregulation of μ-opioid receptors. An inadequate initial opioid response or an inadequate subsequent downregulation could explain the study’s findings. Alternatively, μ-opioid receptors could be located on inhibitory intraneurons, as has been demonstrated in the hippocampus (Drake and Milner 1999). Low binding could reflect less inhibition of interneurons and as a result, the development of a stronger capacity for inhibition during emotional stimulation.
A study that used [carbon-11] carfentanil (11C-carfentanil) to image μ-opioid receptor binding in a priori regions of interest in 16 patients with PTSD, 14 trauma-exposed controls and 15 healthy controls found that both the trauma-exposed control group and PTSD group had lower μ-opioid receptor binding in extended amygdala, nucleus accumbens and dorsal frontal and insular cortex but had higher binding potential in the orbitofrontal cortex. PTSD patients exhibited reduced binding potential in ACC compared to both control groups. Mu-opioid receptor binding in trauma-exposed controls was lower in the amygdala but higher in orbitofrontal cortex compared with PTSD patients and healthy controls (Liberzon et al. 2007b). The authors suggest that trauma exposure leads to widespread changes in the function of the endogenous opioid system, possibly involving downregulation of the limbic forebrain, thalamus and associated cortical regions (insula, ACC, mPFC), along with upregulation of OFC and subgenual ACC receptors. Furthermore, the abnormal responses seen in PTSD may reflect an inability to adequately activate and subsequently downregulate μ-opioid receptors in the amygdala, thalamus and sublenticular extended amygdala and to adequately upregulate these receptors in the OFC and subgenual ACC. Given the role of OFC and subgenual ACC in reward and negative reinforcement and self-induced sadness, these abnormalities may contribute to the anhedonia and emotional numbing observed in PTSD.
14.3.5 Dopaminergic System
In animal models, overstimulation of the dopaminergic system is related to potentiation of fear conditioning (Morrow et al. 1996) and delayed extinction of fear (Borowski and Kokkinidis 1998). Higher levels of dopamine in a stressful situation may consolidate memories related to the traumatic event. Urinary dopamine levels are raised in combat veterans (Yehuda et al. 1992) and in at least some subgroups of children with PTSD (De Bellis et al. 1999). One group (Hamner and Diamond 1993) also demonstrated that patients with PTSD have higher plasma dopamine levels. Genetic studies by several groups have found associations between variants of D2 receptor and PTSD (Comings et al. 1996; Young et al. 2002), between variations in the D4 receptor gene and intensity of PTSD symptoms (Dragan and Oniszczenko 2009), and between variations of the dopamine transporter gene and the likelihood of developing PTSD when exposed to a natural disaster (Drury et al. 2009).
Based on the rationale of a disordered dopaminergic system in PTSD, a study imaging presynaptic dopamine transporter density was conducted using 2-[[2-[[[3-(4-chlorophenyl)-8-methyl-8-azabicyclo[3.2.1]oct-2-yl]methyl](2-mercaptoethyl)amino]ethyl]amino]-ethanethiolato(3-)-oxo-[1R-(exo-exo)]-[technetium-99m] (99mTc-TRODAT-1) SPECT in 21 patients with PTSD and 21 trauma-exposed control subjects. PTSD patients had a higher DAT binding potential in left and right striatum compared to the control group. No clear correlation was found with PTSD symptoms (Hoexter et al. 2012). The authors suggest that their findings support a hypothesis of dopaminergic hyperactivity in PTSD. The authors propose that DAT density increases may not be specifically related to PTSD core symptoms, but rather to the general processing of salient events.
14.3.6 Nicotinic System
Nicotinic acetylcholine receptors (nAChRs) have been implicated in both memory dysfunction and dysregulation of arousal (Gotti et al. 1997; Levin et al. 2006). A study that used a ligand for β2 nicotinic acetylcholine receptors [iodine-123]-5-IA-85380 ([123I]5-IA) to perform SPECT imaging of ten PTSD patients and ten healthy controls reported that in examining a priori regions of interest, relative to never-smoking controls, never-smoking PTSD patients showed higher β2-nAChR availability in the medial temporal cortex. The significance of this difference was weakened when people with prior nicotine exposure were included. A significant association was found between thalamic ligand binding and re-experiencing symptoms of PTSD (Czermak et al. 2008). The authors suggest that their findings support the involvement of β2-nAChRs in PTSD and also speculate that β2-nAChRs contribute to re-experiencing symptoms in PTSD by modulating the sensory input to the cortex (via thalamus).
14.3.7 Hypothalamic-Pituitary-Adrenal (HPA) Axis
The HPA axis is also believed to play a role in PTSD. Through its action on glucocorticoid receptors, cortisol exerts negative feedback on pituitary, hypothalamus and other sites (De Kloet and Reul 1987; Jacobson and Sapolsky 1991), possibly including mPFC (Diorio et al. 1993; Kern et al. 2008). Once the acute stress has been removed and the amygdala is no longer activated by perception of external threat, it activates negative feedback inhibition of the HPA axis in tandem with the hippocampus (McEwen et al. 1987), restoring hormone levels to baseline conditions.
PTSD was initially hypothesised to represent a state of chronic hypercortisolism, but more recent evidence indicates that it is associated with hypocortisolism (Mason et al. 1986; Yehuda et al. 1990b, 1993, 1995a, b; Meewisse et al. 2007). Yehuda (2001) proposes that this low cortisol in PTSD is a downstream manifestation of enhanced negative feedback inhibition resulting from increased glucocorticoid receptor number and sensitivity in the pituitary gland. This is supported by evidence of exaggerated endogenous cortisol suppression with low-dose dexamethasone suppression test in PTSD (Yehuda et al. 1995b) and by the fact that blocking the negative feedback of cortisol on pituitary using metyrapone results in an exaggerated ACTH response in PTSD (Yehuda et al. 1996).
Neuroimaging research provides some support for altered glucocorticoid responsiveness in PTSD. A study that measured changes in brain glucose metabolism following hydrocortisone or placebo administration in 16 patients with PTSD and 16 trauma-exposed controls with 18F-FDG PET found no differences in baseline cortisol levels between the two groups. PTSD patients showed a decrease in activity in the ACC with hydrocortisone, whereas the controls showed an increase – presumably reflecting differences in central glucocorticoid responsiveness. The PTSD group also demonstrated increased metabolism in the right hippocampus and right ventral amygdala in response to hydrocortisone (Yehuda et al. 2009). They found that the net effect of hydrocortisone was to restore a normal inverse association between the ACC and amygdala in the PTSD group but to disrupt this neural network in the control group.
Both animal and human research suggests a role for cortisol in causing hippocampal dysfunction in PTSD, with evidence to suggest that hypercortisolism (Sapolsky et al. 1986, 1990; McEwen 1999) or low cortisol levels (Raison and Miller 2003) are detrimental to hippocampus function and result in volume loss. Alternatively or additionally, altered (increased or decreased) glucocorticoid receptor sensitivity might account for hippocampal damage (Yehuda et al. 2001b; Raison and Miller 2003). One study correlated HPA-axis function, hippocampal volume (measured by MRI), hippocampal function (measured using FDG) and memory tasks in 12 patients with PTSD and 8 trauma-exposed controls. In addition to baseline imaging and neuroendocrine measures, memory tasks and PET imaging were performed (together with ACTH and cortisol measurements) following placebo and hydrocortisone administration. Baseline urinary cortisol excretion was the same in both groups but there was an exaggerated suppression of cortisol with dexamethasone suppression test in the PTSD group. Baseline hippocampal volume asymmetry varied between PTSD and control groups. These differences at baseline were negatively associated with cortisol, ACTH and hemispheric differences in hippocampal metabolism and positively associated with retention, showing an association between structural and functional measures of hippocampal structure, neuroendocrine activity and memory performance. Following hydrocortisone administration, these associations were no longer present. When compared to placebo, the PTSD group demonstrated an increase in hippocampal activation in response to hydrocortisone, whereas the control group showed a decrease. In addition, the PTSD group demonstrated greater suppression of ACTH in response to hydrocortisone than controls. Hydrocortisone did not impact memory performance in PTSD but worsened memory task performance in the control group (Yehuda et al. 2010). The authors concluded that differences in brain metabolic responses may reflect differences in peripheral and central glucocorticoid responsiveness. An earlier study supports this (Yehuda et al. 2009). Nevertheless, not all data are consistent; one study found a negative correlation between endogenous cortisol levels and rCBF in a region most likely representing hippocampal perfusion (Bonne et al. 2003 – see below), and another study found no involvement of hippocampus in PTSD (Liberzon et al. 2007a).
A way of exploring the role of the HPA axis in PTSD is to correlate endogenous cortisol levels with SPECT or PET findings. This was examined in a study in which 11 patients with PTSD, 17 trauma-exposed controls and 11 healthy controls were scanned using 99mTc-HMPAO SPECT. No significant differences were detected between groups in baseline cortisol levels prior to scanning. The group reported a negative correlation between cortisol level and rCBF to medial temporal lobes bilaterally and a positive correlation between cortisol level and fronto-cingulate transitional cortex perfusion in the PTSD group. In the trauma-exposed group, negative correlations between cortisol level and bilateral fronto-cingulate transitional cortex as well as bilateral vmPFC were reported (Bonne et al. 2003). The authors suggested that the contrasting pattern of correlation between cortisol and mPFC/fronto-cingulate transitional cortex in PTSD and trauma-exposed controls may be related to the inadequate shutoff of the stress response in PTSD, while the negative correlation between medial temporal lobe rCBF and cortisol indicates either increased hippocampal control of HPA-axis secretion or a negative effect of cortisol on hippocampal perfusion (since hippocampus is probably the major contributor to medial temporal cortex perfusion measurement).
Similarly, an elegant study conducted to explore the relationship between hypothalamic-pituitary-adrenal axis activity and cortisol release and rCBF, specifically tried to identify circuits that activate the HPA axis and structures that are modulated by cortisol release. The group examined 16 patients with combat-related PTSD, 15 volunteers with combat-exposure but no PTSD and 15 normal controls by performing 15O-water PET imaging during autobiographical trauma-related or neutral event script reading as well as in response to emotional pictures from the International Affective Picture System (Lang and Cuthbert 1997). In the PTSD group there were positive correlations between rCBF and ACTH response in the right insula, rACC and right prefrontal cortex, and unlike trauma-exposed controls, PTSD subjects did not have positive correlations between rCBF and subsequent ACTH response in dmPFC and parahippocampal gyrus (Liberzon et al. 2007a). The implications of these data are that insula and rACC are dysfunctional in PTSD and increases in rCBF in these regions may represent activity in circuits that activate the HPA axis. It is possible that deficient activation of dmPFC (when compared to trauma-exposed controls) in PTSD represents a failure to recruit modulatory circuits. To investigate cortical regions through which cortisol might modulate subsequent cognitive/emotional processing, they examined the relationship between pre-stimulus cortisol level and subsequent cortical responses (rCBF) to traumatic stimulus. They found that only the PTSD group had a positive correlation between pre-stimulus cortisol and a subsequent rCBF response to traumatic stimulus in subgenual ACC, bilateral superior temporal gyrus and right temporal lobe. The authors speculate that this cortisol-rCBF covariation suggests that cortisol may potentially “prime” the circuits that detect emotional cues in auditory stimuli. A reanalysis of the data obtained in the study by Liberzon et al. (2007a) found that trauma recall elicited ACTH responses in some but not all participants, regardless of PTSD status, leading to the conclusion that HPA-axis pathology in PTSD cannot be ascribed solely to a failure of the ACTH response to stressful stimuli (King et al. 2009).
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


