and Aditya G. Shivane1
(1)
Cellular and Anatomical Pathology Level 4, Derriford Hospital, Plymouth, UK
Abstract
Neurodegenerative disorders have become one of the most important group of diseases in terms of disability and socioeconomic consequence to society. Our understanding of the pathogenesis of these disorders and their classification has increased dramatically in the last few years. This chapter describes the classification, pathogenesis and pathology of the different neurodegenerative disorders.
Keywords
DementiaParkinson’s diseaseCerebellar degenerationCJDAlzheimer’s diseaseNeurodegenerative diseases are characterised by progressive neuronal dysfunction and loss, often associated with accumulation of an abnormal protein within the central nervous system (CNS). Most disorders progress over a number of years and currently lack effective treatments. They can be classified in a number of different ways including via the clinical presentation (Fig. 12.1), type of protein that accumulates (Table 12.1), pathological processes and underlying genetic abnormalities.
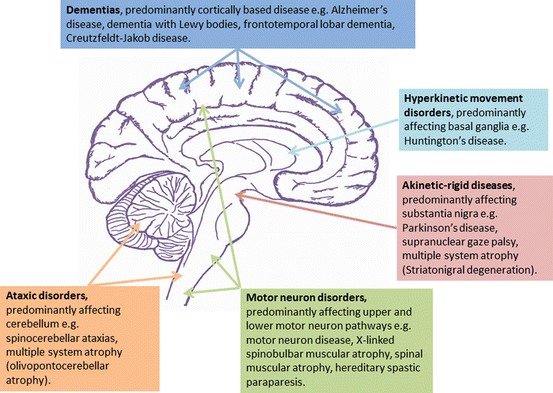
Fig. 12.1
Diagram showing neurodegenerative diseases based on their clinical presentation and area of the nervous system affected
Table 12.1
Neurodegenerative disorders associated with abnormal protein accumulation
Alzheimer’s disease | Aβ and tau |
Familial British dementia | ABri and tau |
Progressive supranuclear palsy | Tau |
Corticobasal degeneration | Tau |
Tangle-only dementia | Tau |
Argyrophilic grain dementia | Tau |
Dementia with Lewy bodies | α synuclein |
Parkinson’s disease | α synuclein |
Multiple system atrophy | α synuclein |
Neurodegeneration with brain iron accumulation | Iron and α synuclein |
Neuroferritinopathy | Ferritin |
Frontotemporal dementias: | |
FTLD-tau | Tau |
FTLD-TDP | TDP43 |
FTLD-FUS | FUS |
FTLD-UPS | Unknown |
FTLD-ni | Unknown |
Motor neuron disease (sporadic) | TDP43 |
Huntington’s disease | Huntingtin |
Creutzfeldt-Jakob disease | Prion |
Diagnosis during life is not always possible with certainty, and autopsy examination of the brain and spinal cord is often required. Autopsies also allow for further detailed study of the pathological processes, which in many cases may extend outside of the CNS, and have led to improved diagnostic classification e.g. frontotemporal lobar dementias.
A number of disorders are associated with accumulation of specific proteins within the CNS, often in an abnormal conformation and/or state of phosphorylation. For example hyperphosphorylated tau accumulates in the form of intraneuronal filamentous tangles (neurofibrillary tangles) in several disorders including Alzheimer’s disease, frontotemporal dementia linked to chromosome 17, tangle-only dementia, progressive supranuclear palsy and corticobasal degeneration. Alpha synuclein accumulates in Parkinson’s disease, dementia with Lewy bodies, some forms of autonomic failure and progressive dysphagia, some neuroaxonal dystrophies, and within glial cells in the different forms of multiple system atrophy. Transactive response DNA binding protein 43 (TDP43) accumulates in motor neuron disease and some subtypes of frontotemporal lobar dementia. Polyglutamine, coded for by the nucleotides CAG, accumulates in the trinucleotide repeat expansion disorders including Huntington’s disease (along with the protein huntingtin) and some forms of spinocerebellar ataxia. Prion protein (PrP) accumulates in both the sporadic and inherited forms of prion disease.
The aetiology of many of the neurodegenerative disorders is unknown and they are probably multifactorial, although genetic factors are implicated in many, either as a pre-disposing factor or in some cases specific abnormality/mutation leading to a familial form of the disorder.
12.1 Dementia
Dementia may be defined as a permanent impairment of higher mental functioning in the presence of normal consciousness, including impairment of memory and other functional domains, such as language, visual spatial skills, emotion, personality or cognition. In some forms, particularly frontotemporal lobar dementias, memory disturbance may occur late, and present with behavioural and language dysfunction. Although neurodegenerative disorders are the commonest cause of dementia, vascular, toxic, metabolic, nutritional, benign tumours and inflammatory/infective disorders can also cause dementia. In the management of patients it is particularly important that reversible causes such as obstructive hydrocephalus, vitamin deficiency, hypothyroidism, neurosyphilis and HIV encephalitis are excluded. As well as the disorders discussed in this section, many of the other neurodegenerative disorders which present with other forms of neurological dysfunction discussed later in this chapter, may also lead to dementia.
12.1.1 Alzheimer’s Disease
This is the commonest cause of dementia, accounting for 70–80 % of cases. It typically presents after the age of 60 with memory disturbance progressing with dysphasia and dyspraxia. After a period of several years patients become immobile and mute. Approximately 10 % of cases are familial, although this is more common in the early onset cases which are associated with mutations in Presenilin 1, Presenilin 2 and β amyloid precursor protein gene (βAPP). Alzheimer’s disease develops in virtually all patients with Down’s Syndrome related to trisomy 21 by the age of 40, due to the dose effect of having three copies of the β amyloid precursor protein gene (which is located on chromosome 21). It should be noted that mutations in the tau gene do not lead to Alzheimer’s disease, providing strong evidence that disorder of amyloid metabolism is of primary importance in this disorder. There are large number of genetic polymorphisms that have been linked to increase risk of Alzheimer’s disease [1], but the apolipoprotein E gene polymorphism is best known, with individuals having the ε4 allele having the greatest risk and ε2 allele, the lowest. Previous head injury is also risk factor for Alzheimer’s disease, and adverse outcome from head injury is also associated with the ε4 allele, which has a number of effects on Aβ metabolism [2].
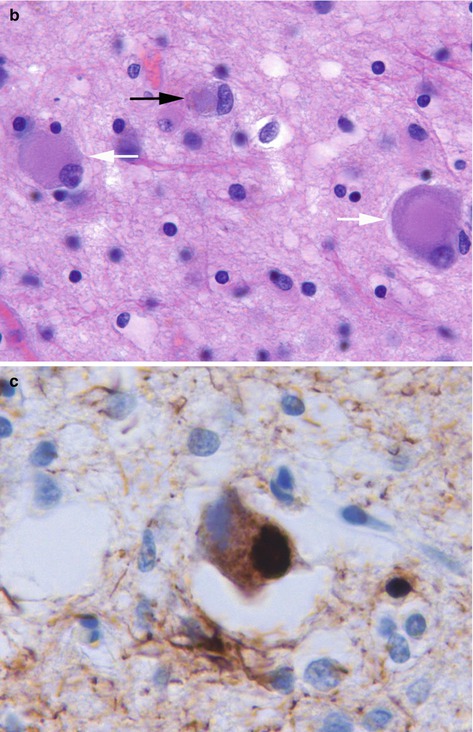
Pathologically the brain shows atrophy (Fig. 12.2), most marked in the medial temporal lobes, and there is relative preservation of the primary motor cortex and occipital lobes. There is accumulation of two proteins, amyloid and abnormally phosphorylated tau. Amyloid accumulates extracellularly in the form of aggregates (plaques) which are composed of the Aβ peptide, a breakdown product of βAPP. Diffuse plaques are common in normal aging, but neuritic plaques (classical senile plaques) are more closely associated with Alzheimer’s disease (Fig. 12.3). In addition, Aβ also accumulates in the small arteries and arterioles within the cerebral and cerebellar cortex and leptomeninges (amyloid angiopathy) (Fig. 12.4). Hyperphosphorylated tau accumulates within neurons diffusely, and in paired helical filaments forming neurofibrillary tangles (Fig. 12.5). Tau also accumulates in the neuropil as small thread-like structures (neuropil threads) and surrounding neuritic plaques, as small bulbous swellings (dystrophic neurites). An early pathological change is loss of synapses, and other common microscopic abnormalities include loss of neurons from the cortex, granulovacuolar degeneration of the pyramidal neurons within the hippocampus and actin accumulation within hippocampal neurons (Hirano bodies).
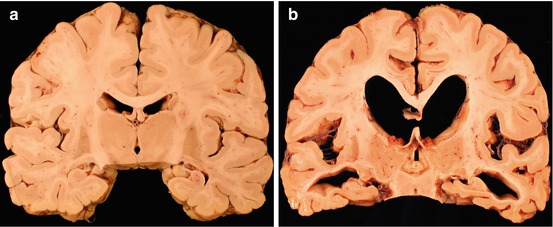
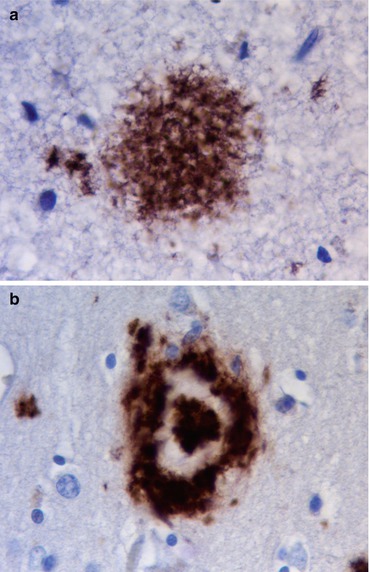
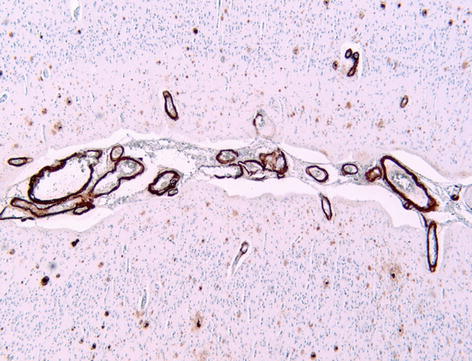
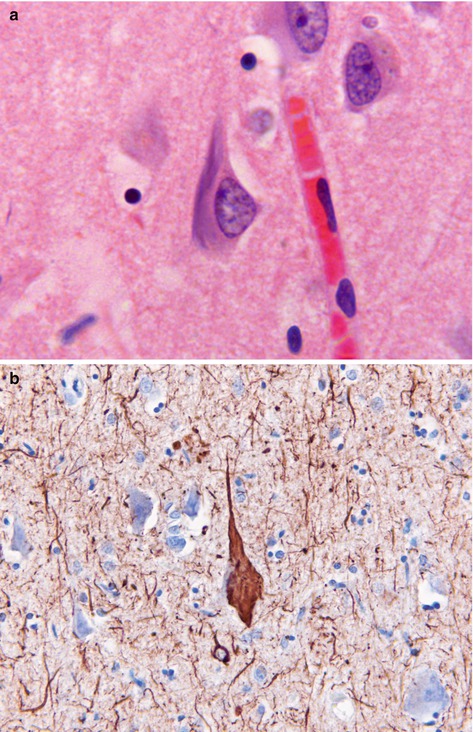
Fig. 12.2
(a) Coronal slice of normal brain. (b) Alzheimer’s disease showing generalised cortical atrophy, most marked in the medial temporal lobes with enlargement of ventricles
Fig. 12.3
(a) Diffuse ‘unstructured’ plaque, common in normal aging. (b) Neuritic plaque, characteristic of Alzheimer’s disease. Immunohistochemistry for A4 protein
Fig. 12.4
Amyloid angiopathy, showing deposition within leptomeningeal and superficial cortical blood vessels, in a patient with Alzheimer’s disease. Immunohistochemistry for A4 protein
Fig. 12.5
(a) Neurofibrillary tangle seen as bundles of filamentous basophilic stained material in the cytoplasm of a neuron. H&E stain. (b) Tau deposition in neuropil as ‘threads’ and within neuronal tangle. Immunohistochemistry for phosphorylated tau protein
Neuronal loss is associated with neurochemical alterations, in particular loss of neurons from the nucleus basalis of Meynert leads to marked reduction of acetylcholine in the cerebral cortex, depletion of the dorsal raphe nucleus and locus ceruleus results in reduced 5 hydroxytryptamine and noradrenaline input into the cortex respectively.
The severity of the histological changes can be classified using either the density and distribution of tangles within the brain (Braak stage – I to VI) [3, 4], or by the density of amyloid plaques (CERAD score – mild, moderate or frequent) [5]. The correlation between the severity of the pathology and of dementia is not absolute, and there are cases with moderately severe Alzheimer’s disease pathology with minimal cognitive impairment [6].
In some elderly patients (particularly women) with dementia the only pathological change may be the presence of neurofibrillary tangles and dystrophic neurites, principally in the medial temporal lobe and substantia nigra (tangle-only dementia).
Another condition which presents with dementia in the elderly, argyrophilic grain dementia, is characterised by small granular and filamentous tau deposits in the hippocampus and other medial temporal lobe structures.
12.1.2 Dementia with Lewy Bodies
This is the second commonest neurodegenerative cause of dementia, accounting for 10–20 % of cases in hospital-based series. The clinical presentation overlaps with that of Alzheimer’s disease, although features such as pronounced fluctuation in symptoms, well-formed visual hallucinations, parkinsonian features and neuroleptic sensitivity are helpful pointers. The pathology overlaps with that of Parkinson’s disease and distinguishing the disorder from Parkinson’s disease with dementia is based upon the presentation, with the latter being defined as dementia occurring at least a year after the presentation of a pure motor Parkinson’s disease.
The brain shows atrophy, particularly in the temporal lobes and microscopically many cases have varying degrees of Alzheimer’s disease type-pathology. However, in addition there are Lewy bodies (Fig. 12.6) and Lewy neurites, including within the limbic system and insular cortex. Lewy bodies and neurites are characterised by accumulation of α synuclein. Lewy body pathology may also be seen in the brain stem. In some cases there may be areas of laminar microvacuolation, resembling the spongiform changes seen in Creutzfeldt-Jakob disease. Severe involvement of the basal nucleus of Meynert leads to cholinergic deficiency, and neuronal loss from the substantia nigra leads to dopamine deficiency.
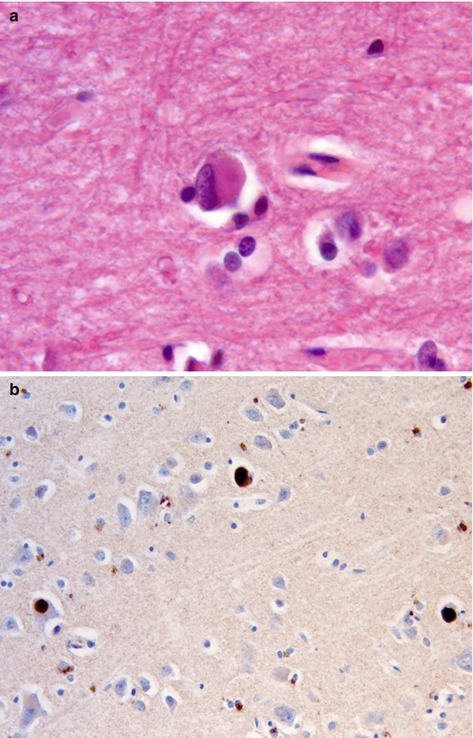
Fig. 12.6
(a) Cortical Lewy body seen as eosinophilic rounded structure within a cortical neuron. H&E stain. (b) Lewy bodies are composed of α synuclein. Immunohistochemistry for α synuclein
Lewy body disorders can be classified pathologically into brain stem predominant (Parkinson’ disease, Lewy body dysphagia) or limbic, neocortical and amygdala predominant forms, using consensus criteria [4].
12.1.3 Frontotemporal Lobar Degeneration
Frontotemporal lobar degeneration (FTLD) accounts for 10–20 % of cases of dementia and may present as either a behavioural variant with changes in social and personal conduct, progressive non-fluent aphasia with an expressive dysphasia, or as semantic dementia with impairment of verbal and visual memory. Some patients may also have motor neuron disease or parkinsonian features. Overall about 50 % of cases of frontotemporal dementia are familial, with an autosomal dominant pattern of inheritance. These dementias may be classified according to the type of protein accumulation (Table 12.1).
FTLD-tau is seen in the chromosome 17-linked dementia, associated with mutations in the tau gene, and in Pick’s disease, which is largely sporadic, and associated with swollen neurons (Pick’s cells) and rounded intracellular neuronal inclusions (Pick bodies) (Fig. 12.7), both of which are associated with tau accumulation. FTLD-TDP is the most common subtype and is seen in cases of dementia associated with sporadic motor neuron disease and in association with mutations in progranulin or valosin containing protein (VCP) genes. Patients may have Paget’s disease and inclusion body myopathy. An expansion of the hexanucleotide repeat in the C9orf72 gene on chromosome 9 is associated with either motor neuron disease or dementia with FTLD-TDP pathology, and is a common cause of familial FTLD. In a small number of cases of FTLD there may be accumulation of fused in sarcoma protein (FUS), ubiquitin-only immunoreactive changes (FTLD-UPS), some of which are associated with mutations in the charged multivesicular body protein 2B (CHMP2B). A few cases of FTLD have no identifiable inclusions (FTLD-ni).