Neurofibromatosis Type 1 and Tuberous Sclerosis Complex
Kaleb H. Yohay
Martha B. Denckla
Neurofibromatosis type 1 (NF1) and tuberous sclerosis complex (TSC) are autosomal dominant genetic conditions that affect multiple organ systems. Although both are tumor syndromes, the pathology related to both disorders extends far beyond the development of tumors. A significant amount of the morbidity associated with these two disorders results from their effects on cognition, behavior, and attention.
Neurofibromatosis Type 1
Neurofibromatosis type 1, also known as von Recklinghausen disease, is a common genetic disorder that affects multiple organ systems. It is inherited in autosomal dominant fashion and has an incidence of about 1 in 3,000 (1). Penetrance is 100% although there is wide variation in phenotype severity (2). Mosaicism may also occur. The NF1 gene is located on chromosome 17q11.2 and encodes for a protein called neurofibromin (3). Neurofibromin belongs to a family of proteins called GTPase activating proteins (GAP), which downregulate the proto-oncogene p21-RAS.
The diagnosis of NF1 is based on the presence of clinical features (Table 10.1). NF1 is usually diagnosed in infancy or early childhood. Essentially all patients with NF1 meet the criteria by young adulthood, with more than 95% meeting clinical criteria by age 8. About half of children with sporadic NF1 will meet the diagnostic criteria by age 1 (4). Genetic testing for NF1 is available but is of limited utility in the clinical setting and is generally reserved only for equivocal cases. Mutation analysis is not helpful in predicting phenotype or prognosis, with the exception of cases in which the entire gene is deleted, which may predict more severe disease (5).
Clinical Features
CAFÉ-AU-LAIT MACULES AND FRECKLING
Café-au-lait macules (CALs) are the most consistent manifestation of NF1 and are typically the first diagnostic criteria met by patients with NF1. These hyperpigmented macules are often present at birth and generally appear within the first year of life (4). They may increase in both size and number with age, although later in adulthood may fade.
CALs are common in the normal population, with approximately 25% of the general population having one to three CALs. Greater than 95% of all patients with NF1 have CALs. CALs are generally oval with relatively smooth borders (Fig. 10.1). They usually range in size between 1 and 4 cm. Larger, more irregularly shaped CALs can be seen in NF1 but may also be indicative of other disorders associated with the presence of CALs such as McCune-Albright syndrome.
CALs are common in the normal population, with approximately 25% of the general population having one to three CALs. Greater than 95% of all patients with NF1 have CALs. CALs are generally oval with relatively smooth borders (Fig. 10.1). They usually range in size between 1 and 4 cm. Larger, more irregularly shaped CALs can be seen in NF1 but may also be indicative of other disorders associated with the presence of CALs such as McCune-Albright syndrome.
| ||||||||||||||||||
Axillary or inguinal freckling is also a diagnostic criterion for NF1. Freckling in NF1 is distinct in that it appears in non-sun-exposed areas such as the armpits, the groin area, or other intertriginous areas (Fig. 10.2). The freckling generally appears during childhood. Older patients with NF1 may develop more diffuse hyperpigmentation and freckling.
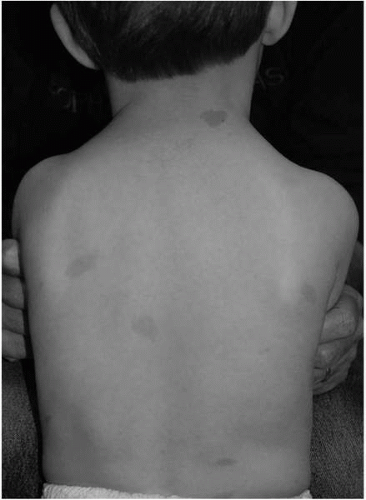 ▪ FIGURE 10.1 Café-au-lait macules (CALs). Typical appearance of CALs in a 4-year-old boy with NF1. They are typically ovoid and smooth bordered. |
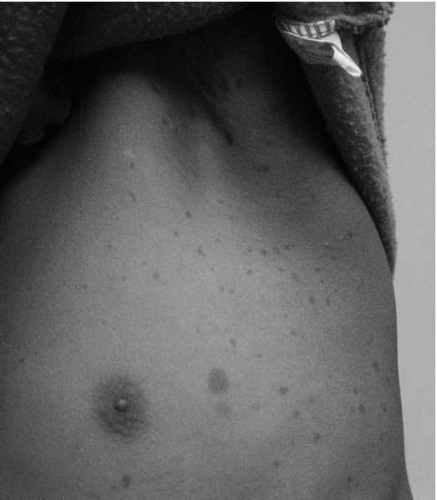 ▪ FIGURE 10.2 Axillary freckling. Typical appearance of axillary freckles in an 8-year-old girl with NF1. |
NEUROFIBROMAS
Neurofibromas are the defining characteristic of NF1. Neurofibromas can be divided into several subtypes: cutaneous neurofibromas, subcutaneous neurofibromas, nodular plexiform neurofibromas, and diffuse plexiform neurofibromas. All types are composed of the mix of Schwann cells, fibroblasts, and mast cells (6). Cutaneous neurofibromas are soft, fleshy tumors that arise from the peripheral nerve sheath at the skin’s surface (Fig. 10.3). They generally appear during late childhood or young adulthood (4). They can vary in color, size, and density. Some are sessile, whereas others are pedunculated. They are not premalignant but are often cosmetically significant and may cause itching or, less commonly, pain. Subcutaneous neurofibromas are generally firm, often tender nodules that arise along the course of the peripheral nerve below the surface of the skin. They also generally appear during adolescence or young adulthood. Like cutaneous neurofibromas, these are not considered to be premalignant but may become cosmetically significant or
cause discomfort. Nodular plexiform neurofibromas arise along the proximal nerve roots and, because of their proximity to the spinal cord, can cause cord compression as well as vertebral erosion and scoliosis. Diffuse plexiform neurofibromas often involve multiple nerve fascicles or long lengths of nerve fibers. These are thought to be congenital and their origin may not be apparent initially (Fig. 10.4). Plexiform neurofibromas can be particularly problematical, in that they can become quite large, causing disfigurement or functional impairment or impingement on vital organs or structures.
cause discomfort. Nodular plexiform neurofibromas arise along the proximal nerve roots and, because of their proximity to the spinal cord, can cause cord compression as well as vertebral erosion and scoliosis. Diffuse plexiform neurofibromas often involve multiple nerve fascicles or long lengths of nerve fibers. These are thought to be congenital and their origin may not be apparent initially (Fig. 10.4). Plexiform neurofibromas can be particularly problematical, in that they can become quite large, causing disfigurement or functional impairment or impingement on vital organs or structures.
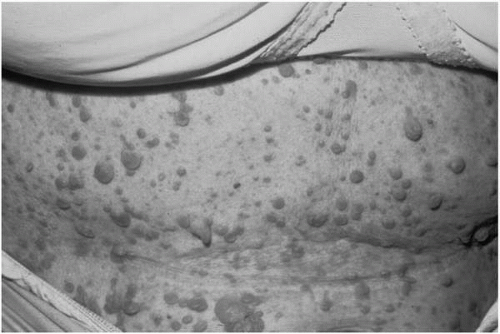 ▪ FIGURE 10.3 Cutaneous neurofibromas. Cutaneous neurofibromas are soft, fleshy benign growths arising from intracutaneous nerves. They can be sessile or pedunculated and can vary markedly in color, size and density. (From Goodheart HP, MD. Goodheart’s Photoguide of Common Skin Disorders. 2nd ed. Philadelphia: Lippincott Williams & Wilkins; 2003; with permission.) |
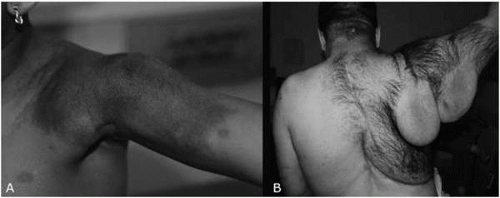 ▪ FIGURE 10.4 A: Plexiform neurofibroma in the shoulder of a 4-year-old girl with neurofibromatosis type 1 (NF1). Underlying the area of rough, hyperpigmented skin is a palpable, heterogeneous, mobile, painless mass. B: Plexiform neurofibroma in a 38-year-old man with NF1. |
Currently, no effective medical treatments for neurofibromas are available. Cutaneous neurofibromas may be removed with traditional plastic surgical techniques or by laser ablation. However, removal of cutaneous neurofibromas is generally reserved for those that cause discomfort or are cosmetically significant. Plexiform neurofibromas are particularly difficult to treat because they are generally large, irregular, and highly vascular and often involve numerous nerves. As a result, complete resection is generally not achievable and surgery is reserved for situations in which there is significant functional impairment, severe discomfort, or disfigurement. Furthermore, their growth patterns are variable, which makes treatment choices more difficult. These masses may stay quiescent throughout life, causing little or no morbidity, or may go through periods of rapid growth. They also have the potential for malignant transformation to a malignant peripheral nerve sheath tumor (MPNST). The lifetime risk of malignant transformation is probably between 5% and 13% (7,8).
CENTRAL NERVOUS SYSTEM TUMORS
Optic pathway gliomas (OPGs) are the most common central nervous system (CNS) tumor in patients with NF1. OPGs occur in up to 15% of patients with NF1 and generally develop during the first decade of life (9,10). These tumors are histologically low-grade astrocytomas arising anywhere along the optic pathway (Fig. 10.5). Most optic pathway gliomas are asymptomatic; however in the one third that are symptomatic, patients typically present with vision loss or proptosis or both. They may also present with signs of early onset of puberty if the tumor originates near the optic chiasm. Most patients with NF1 and optic pathway gliomas are managed conservatively with serial neuroimaging and ophthalmologic evaluation. For patients with progressive symptomatic disease, treatment with chemotherapy and, less commonly, surgery or radiation may be considered.
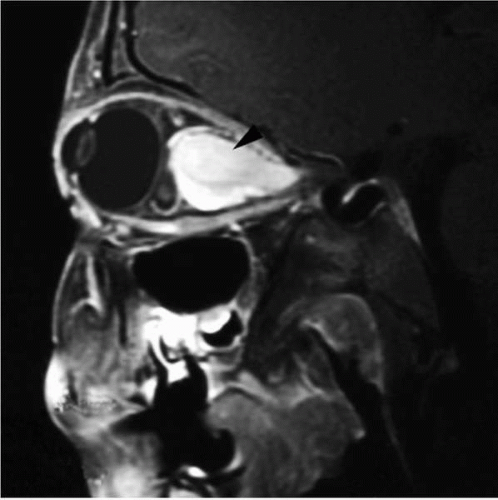 ▪ FIGURE 10.5 Optic pathway glioma in a 5-year-old boy with neurofibromatosis type 1 (NF1). This sagittal magnetic resonance imaging (MRI) of the orbits demonstrates an avidly enhancing mass (arrowhead) within the optic nerve displacing the globe forward. |
Patients with NF1 have an elevated risk for the development of brainstem gliomas. Unlike brainstem gliomas in the non-NF1 population, they are typically slow growing and asymptomatic and may not require treatment (11,12). There may also be an increased risk of development of other intracranial tumors, including low- and high-grade astrocytomas, medulloblastomas, meningiomas, ependymomas, and malignant schwannomas of the cranial nerves (13,14).
NON-CENTRAL NERVOUS SYSTEM MALIGNANCY
The most common malignancies seen in patients with NF1 are MPNSTs. These tumors are very aggressive, often fatal sarcomas. The lifetime risk of the development of a MPNST is between 5% and 13% (7,8). Typically, malignant transformation of the plexiform neurofibroma is heralded by rapid, asymmetrical growth of a known plexiform neurofibroma and new onset of significant pain. Imaging with magnetic resonance imaging (MRI) or positron emission tomography (PET) may be helpful in diagnosing MPNSTs. Treatment is difficult and generally requires a combination of surgical resection and chemotherapy.
Patients with NF1 are at low but increased risk of development of several other non-CNS malignancies, including juvenile chronic myelogenous leukemia, pheochromocytoma, and rhabdomyosarcoma.
OCULAR MANIFESTATIONS
The most common eye finding in patients with NF1 is the presence of Lisch nodules. These are small, pigmented, raised hamartomas present on the iris. These are best visualized using a slit lamp examination but may be seen through direct visualization. Lisch nodules generally cause no apparent symptoms. Congenital glaucoma is occasionally seen in NF1.
SKELETAL MANIFESTATIONS
Patients with NF1 are at increased risk for development of several bony lesions, including sphenoid wing dysplasia, pseudarthrosis, vertebral dysplasias, and scoliosis. Sphenoid wing dysplasia is a congenital abnormality of the sphenoid bone of the skull. This may be a disfiguring complication and may occur with or without the presence of an associated plexiform neurofibroma. It may result in intrusion of the temporal lobe into the orbit, causing a pulsatile proptosis. Pseudarthrosis, or false joint, may result from thinning of the long bone cortex, most commonly of the tibia, followed by pathological fracture and poor bone regrowth and healing. Pseudarthroses most typically manifest prior to the age of 2 years. Scoliosis occurs in more than 10% of patients with NF1. It may be a result of deformation of the vertebral bodies by nodular plexiform neurofibromas in the vertebral foramina or from dysplasia of the vertebral bodies.
CARDIOVASCULAR ABNORMALITIES
Patients with NF1 are also at increased risk of congenital and acquired cardiovascular abnormalities. These include vasculopathy, hypertension, and a wide range of congenital heart defects. Symptomatic vasculopathy is relatively uncommon but may result in stenosis, inclusion, aneurysm formation, or fistula formation. The most common symptomatic lesions are renal artery stenosis, leading to hypertension or stenosis of the internal carotid arteries, or middle cerebral artery stenosis, resulting in stroke, brain hypoperfusion, or seizures.
Hypertension may be seen in children with NF1 most commonly as a result of renal artery stenosis. Adult patients with NF1 may develop hypertension as a result of the development of pheochromocytoma, primary hypertension, or, less commonly, renal artery stenosis.
OTHER CENTRAL NERVOUS SYSTEM MANIFESTATIONS
Seizures are a relatively uncommon manifestation of NF1 with a prevalence of approximately 4%. They may be focal or generalized and may begin at any age. Macrocephaly occurs in approximately one half of children with NF1. Most commonly, macrocephaly is due to increased brain volume. Less commonly, it is due to hydrocephalus from aqueductal stenosis.
Unidentified bright objects (UBOs) are characteristic lesions in the brain seen on MRI in patients with NF1 (Fig. 10.6). These lesions most likely represent areas of myelin vacuolization and are not thought to be malignant or premalignant. They have no mass effect or contest enhancement on neuroimaging. They are generally not associated with focal neurological deficits; however, it has been suggested that the location or number of UBOs may be correlated with the degree of cognitive and behavioral dysfunction.
COGNITIVE AND BEHAVIORAL MANIFESTATIONS
Behavioral and cognitive manifestations are common in NF1 and represent a major source of morbidity. Intellectual development, learning disabilities, psychosocial impairments, attention problems, and emotional difficulties have all been associated with NF1.
Overall, intelligence appears to be somewhat affected in the NF1 population. The incidence of mental retardation in patients with neurofibromatosis is quite low with approximately 4% to 8% of patients having intelligence quotients (IQs) less than 70 (15). However, numerous studies have repeatedly shown that although patients with NF1 have IQs within the normal range, there is a downward shift in IQ of between 5 and 10 points when compared with unaffected siblings (16, 17, 18). There does not seem to be a consistent pattern in differences between the verbal IQ and performance IQ (19, 20, 21, 22, 23).
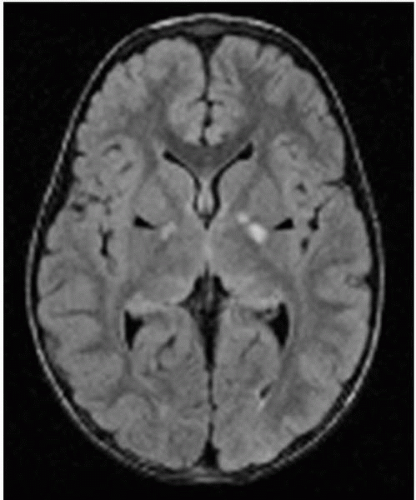 ▪ FIGURE 10.6 Unidentified bright objects (UBOs) in a patient with neurofibromatosis type 1 (NF1). This axial magnetic resonance imaging (MRI) [FLuid Attenuated Inversion Recovery (FLAIR) sequence] shows T2 bright lesions in the internal capsule bilaterally (black arrowheads). These do not enhance and cause no mass effect, typical of UBOs. |
LEARNING DISABILITIES AND COGNITIVE PHENOTYPE
The prevalence of learning disabilities in children with NF1 is clearly increased with rates reported between 20% and 65% (24, 25, 26, 27, 28, 29, 30), compared with approximately 10% in the general population (30a). Specific patterns of learning disability have long been postulated, in particular that the cognitive phenotype of NF1 is primarily characterized by the presence of nonverbal learning disabilities. Over two decades ago Eliason (30b) demonstrated a high incidence of visual perceptual disability in children with NF1 (56%). Studies comparing children with NF1 to their unaffected siblings have demonstrated a leftward shift of the IQ and increased incidence of subtle neurological signs and visuospatial dysfunction (31, 32, 33). In particular, the Judgment of Line Orientation test, a measure of visuospatial function, is consistently abnormal in children with NF1 and learning disabilities (34). Other studies have demonstrated poor performance on tests of spatial memory (35) and visual motor integration (36). However, other work suggests that children with NF1 and learning disabilities demonstrate a broad range of cognitive deficits, such that the concept of a cognitive phenotype may not be useful. More recent studies have demonstrated that verbal learning disabilities may be as common as nonverbal learning disabilities (37, 38, 39, 40, 41, 42) and may include impairments in phonological awareness, expressive and receptive vocabulary, verbal reasoning, and academic achievement in reading and writing (38). Furthermore, deficits in attention and organizational skills have also been demonstrated (43, 44, 45).
Attention deficit hyperactivity disorder (ADHD) also occurs with increased frequency in the NF1 population. In one study of children with NF1, 42% met criteria for ADHD, compared with 13% of their unaffected siblings (46). In another study of 81 children with neurofibromatosis, 38% met criteria for ADHD compared with 12% in their unaffected siblings. Furthermore, 46% of children with NF1 and a specific or global learning delay met criteria for ADHD (47). Children with NF1 and ADHD appear to respond to treatment with stimulants similarly to children having ADHD without NF1 (48).
Research examining the relationship between NF1 and other psychiatric disturbances such as anxiety and depression is quite limited, although some preliminary results suggest an association (49).
The underlying pathogenic mechanisms for the cognitive and behavioral aspects of NF1 are not well understood. In addition to its role as a tumor suppressor, neurofibromin may also play a role in embryonic CNS development (50,51). Dysfunction of neurofibromin may therefore result in underlying brain structural abnormalities and cognitive dysfunction; however, neuropathological data to support or refute this are scarce (52). Neuroimaging has provided the best tool for identifying structural and cognitive correlates. Some studies have suggested an association with UBOs and cognitive deficits. UBOs occur in 60% to 70% of children younger than 16 years with NF1, although they often disappear later in adulthood (53,54). The relationship between UBOs and cognition is complex. The presence of UBOs during childhood has been shown to be a predictor of cognitive dysfunction during adulthood (55). The number of locations UBOs occupy may correlate more with cognitive function than the total number or volume of UBOs (56). Location of the UBOs may also be important. One study demonstrated that thalamic lesions were associated with poorer cognitive function (57). Interestingly, magnetic resonance spectroscopy reveals abnormalities in the thalami of patients with NF1. These abnormalities are most pronounced in patients with UBOs in the thalami but were observed to be also present to a lesser extent in patients without UBOs in the thalami (58). This certainly suggests that UBOs may be only a small visible part of the underlying brain pathology present in NF1. Megalencephaly, which may be the result of both gray and white matter volumes (59), has also been suggested to possibly correlate with some aspects of cognitive dysfunction (60).
Related posts:
 Neuroradiological Imaging in Children
Neuroradiological Imaging in Children
 Functional Neuroimaging in Pediatric Populations
Functional Neuroimaging in Pediatric Populations
 Challenges of Congenital Hearing Loss
Challenges of Congenital Hearing Loss
 Developmental Characteristics of Sleep and Sleep Disorders in Children
Developmental Characteristics of Sleep and Sleep Disorders in Children
 Neurobehavioral Aspects of Traumatic Brain Injury in Children
The Diagnostic Approach to Neuropsychiatric Presentations
Neurobehavioral Aspects of Traumatic Brain Injury in Children
The Diagnostic Approach to Neuropsychiatric Presentations
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


