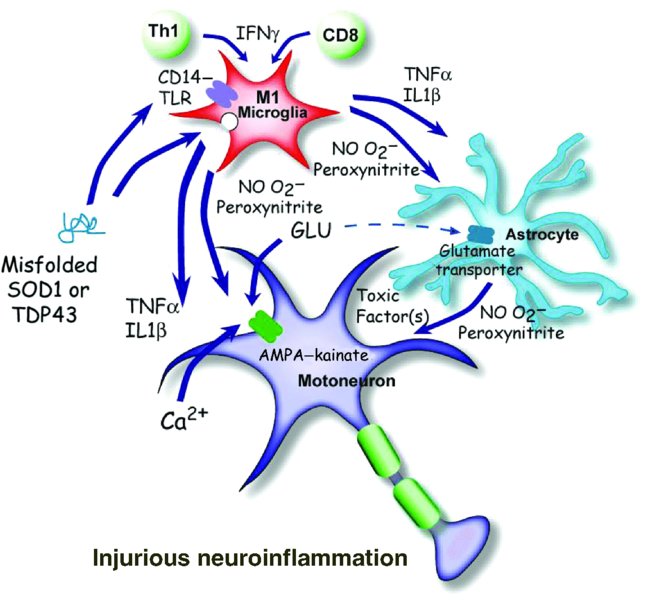
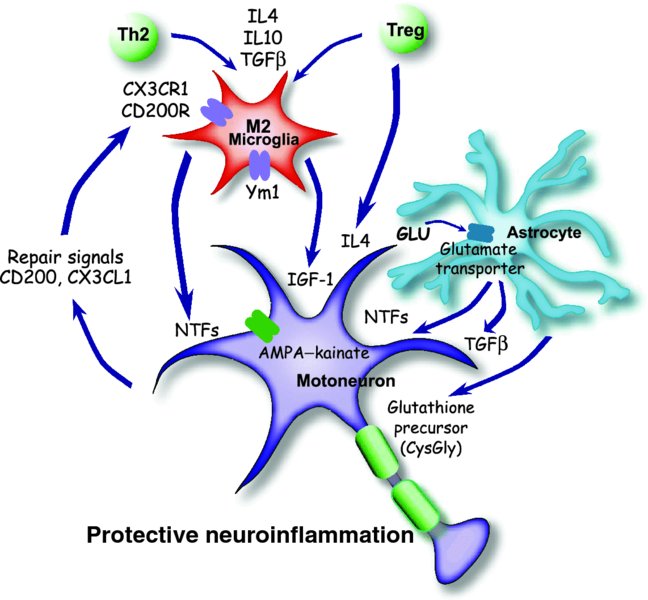
8 Jenny S. Henkel*, David R. Beers, Weihua Zhao and Stanley H. Appel Department of Neurology, Houston Methodist Neurological Institute, The Houston Methodist Hospital Research Institute, The Houston Methodist Hospital, Houston, Texas, USA Amyotrophic lateral sclerosis (ALS), first described by Jean-Marie Charcot in the 19th century and now colloquially referred to as Lou Gehrig’s disease, is a member of a group of adult-onset anterior horn disorders that cause selective, rapidly progressing and irreversible neurodegeneration of the patient’s upper and lower motoneurons (Naganska and Matyja, 2011). The motoneurons in the cortex, brainstem and spinal cord die, resulting in various degrees of weakness, limb musculature atrophy, spasticity, dysarthria (slurred speech) and dysphagia (difficulty with swallowing); the neuromuscular system collapse caused by this devastating disorder ultimately culminates in respiratory failure and death, usually within 4 to 6 years of symptom onset. ALS initiates focally and then spreads to neighbouring structures (Ferraiuolo et al., 2011). Prior to motoneuron cell body loss in the spinal cord, denervation at the neuromuscular junction is observed, followed by a ‘dying back’ of distal branches (Fischer et al., 2004). At autopsy, an obvious loss of motoneurons in the central nervous system (CNS) is evident, while the remaining neurons contain numerous inclusions comprising misfolded proteins, swelling of the perikaryon and proximal axon, mitochondria swellings, vacuoles and neurofilament accumulations. In general, this accumulation of intracellular or extracellular misfolded proteins in the CNS is a common feature of neurodegenerative disorders. In addition to the well-described motoneuron pathologies, the surrounding glia are also affected; proliferating microglia and astrocytes are highly activated and contain numerous inclusions. The worldwide incidence of ALS is approximately 2 per 100,000 individuals, and is fairly uniform except for a few high-incidence regions such as the Kii Peninsula and Guam (Logroscino et al., 2010). The mean age of onset is between 55 and 60 years of age, and it commonly affects more men than women until after age 70, when the ratio approaches 1 to 1. ALS was traditionally considered to be a pure motoneuron disorder (MND); however, recent findings in subsets of patients have highlighted the involvement of the frontal and temporal lobes and, to a lesser extent, sensory and spinocerebellar pathways, as well as the substantia nigra and hippocampal dentate gyrus. Thus, ALS is now regarded as a multisystem disorder in which motoneurons tend to be affected the earliest and the most severely. The pathogenic processes underlying ALS are multifactorial and, at present, not fully understood. Most forms of ALS are idiopathic, having no obvious genetic basis for the disease, and are known as sporadic ALS (sALS), whereas approximately 10% of patients have a heritable form of ALS, referred to as familial ALS (fALS) (Andersen and Al-Chalabi, 2011). In sALS, recent studies have documented the presence of hexanucleotide repeats in chromosome 9, open reading frame 72 (C9ORF72) in 6–10% of the cases (DeJesus-Hernandez et al., 2011; Renton et al., 2011). Several other, less common mutations have been described in non-overlapping lists of candidates that include Ataxin-2 and the Unc-13 vesicular protein. It is likely that sALS, a phenotypically variable syndrome, is a genetically heterogeneous disease with multiple genes initiating motoneuron degeneration through separate but convergent biological pathways. Thus, definitive diagnosis of ALS relies upon a combination of primary clinical findings, systematic inclusion and exclusion of potential genetic factors, and environmental explanations. Unfortunately, the present therapies are mainly symptomatic and fail to halt disease progression; Riluzole, the first and only US Food and Drug Administration (FDA)-approved medication for ALS, only modestly prolongs survival. The absence of an effective treatment can be explained in part by the complex and heterogeneous genetic, biochemical and clinical features of ALS. While ALS accounts for the majority of the motoneuron diseases, the recognition of disease variants and mimic syndromes may lead to further insights into possible causes for the generality of ALS. From a biochemical perspective, the process of motoneuron degeneration is complex and the multifactorial influences and potential biomarkers of ALS have never been assessed in the light of the clinical heterogeneity of ALS. Approximately one-tenth of ALS cases are hereditary, but even within this subset there are currently 13 confirmed Mendelian gene mutations encoding proteins in disparate pathways that appear to be minimally interconnected; however, the genetic penetrance of these genes is not 100%. More significant is the fact that most of the genes associated with these fALS cases code for proteins involved in cellular mass transport (either axonal transport or vesicle trafficking; i.e. spatacsin or vesicle-associated membrane protein (VAMP)); abrogating or reducing cellular oxidative stress (superoxide dismutase); RNA transcription, processing and function (angiogenin, TAR DNA binding (TDP43) and fused in sarcoma (FUS)); endosomal trafficking (alsin and FIG4 homolog); and protein degradation pathways (valosin-containing protein and ubiquitin-2). Interestingly, these genes often code for proteins whose mutation or malfunction results in macromolecular cellular aggregates. Most importantly, in 30–50% of all fALS cases, there is an uncharacterized gene product of C9ORF72 that codes for a protein involved with RNA splicing and formation of nuclear RNA foci. The first identified gene accounting for 20% of the known mutations in fALS is the SOD1 gene encoding the Cu2+–Zn2+ superoxide dismutase 1 (SOD1) enzyme (Rosen et al., 1993). Mutations in SOD1 (mSOD1) account for approximately 20% of the familial cases and were first associated with fALS in 1993. To date, over 165 mutations have been identified in SOD1 that induce disease by a toxic gain of function and not by a loss of enzymatic activity. The most common cause of fALS is the recently identified expanded GGGGCC repeat that is located on 9p21 in the noncoding region of C9ORF72 and accounts for approximately 30–50% of all familial cases. C9ORF72 is a gene that is highly conserved across species and that encodes an uncharacterized protein with no known function. The hexanucleotide repeat expansion prevents expression of the C9ORF72 transcript variant 1 from the mutant allele; however, the defect may also impair RNA processing in general. While little is known about the C9ORF72-encoded protein, it appears to be expressed in the regions of affected neurons in cytoplasmic and synaptic localizations; the proposed C9ORF72 protein is structurally related to DENN domain proteins, which are highly conserved guanosine diphosphate (GDP)–guanosine triphosphate (GTP) exchange factors for Rab GTPases (Levine et al., 2013). The mechanism of disease mediated by the GGGGCC repeat is still unresolved, but it has been attributed to haplo-insufficiency, a toxic gain of function or the synthesis of toxic di-peptide repeat proteins (Renoux and Todd, 2012). With regard to the di-peptide hypothesis, a repeat associated non-ATG translation (RAN translation) across expanded GGGGCC repeats, an unconventional mode of translation in the absence of an initiating ATG, may produce three two-amino-acid alternating copolymers – (glycine–alanine)n, (glycine–arginine)n and (glycine–proline)n – similar to the polyglutamine, polyserine or polyalanine tracts found with expanded CAG repeats. High-molecular-weight insoluble accumulations of poly-(glycine–proline) peptides are detected, with little or no poly-(glycine–arginine) or poly-(glycine–alanine) peptides, in brain homogenates of C9ORF72 ALS patients with frontotemporal dementia; poly-(glycine–proline) peptides appear specific for C9ORF72 ALS because none of the three possible two-amino-acid copolymers were found in the CNS of patients with other neurodegenerative diseases (Ash et al., 2013). However, it remains to be determined whether the polypeptides generated by RAN translation are neurotoxic. The third most common cause of fALS involve mutations in the related genes TARDBP and FUS encoding the DNA- and RNA-binding proteins, TDP43 and FUS–translocated in liposarcoma (TLS), respectively (Mackenzie et al., 2010). Mutations in these proteins, which are associated with fALS, were first identified in 2008 and 2009, respectively. Since their initial discovery, many other mutations in TARDBP and FUS have been found and comprise approximately 4–5% of all fALS cases. Although ALS has been traditionally viewed as a motoneuron cell-autonomous disorder, the current dogma suggests that ALS is a non-cell-autonomous neurodegenerative disorder, in which neurons do not die alone, but numerous cell types (both neuronal and non-neuronal) play a role in disease initiation and progression. There is now the emerging and generally accepted concept that both the innate and adaptive immune systems play early and important neuroinflammatory roles at the neuromuscular junction and the distal axonal compartment, and in the CNS compartment, in the disease pathophysiology of both fALS and sALS; there is an increasingly recognized role of macrophages, neighbouring CNS microglia and infiltrating lymphocytes and their particular molecular signals, in motoneuron survival and cell death. Even though the CNS has traditionally been considered immunologically privileged, and in light of this emerging concept, there has been a re-evaluation of this tenet and neuroinflammation is now recognized to be a prominent feature of many classic neurodegenerative disorders, including ALS. Furthermore, denervation at the neuromuscular junction is observed prior to motoneuron cell body loss in the spinal cord, suggesting that motoneuron pathology begins at the distal axon and proceeds in a ‘dying back’ process. Thus, in ALS and other neurodegenerative disorders, microglial activation and T cell infiltration are pathological hallmarks at sites of neuronal injury, yet such neuroinflammation has been considered the consequence and not the cause of neuronal injury; both innate and adaptive immune systems respond to, as well as contribute to, the pathology and tissue destruction (see Figure 8.1). However, more recent evidence challenges this belief and suggests that at an early phase of disease, the immune system may contribute to neuroprotective functions (see Figure 8.2). Figure 8.1 An injurious neuroinflammatory response. M1 microglia, ROS and aberrant astroglia contribute to motoneuron injury. Mutant or misfolded SOD1 or TDP43 proteins interact with CD14–TLR, inducing M1 microglia, secretion of proinflammatory cytokines and ROS. ROS increase the number, or Ca2+ permeability, of AMPA receptors and reduce glutamate transporter function on astrocytes, allowing excessive activation of AMPA receptors. Astrocytes have reduced trophic factors and up-regulate the release of toxic factors. The result of this is a transformation of microglia from an M2 to an M1 phenotype, which promotes neurotoxicity. Figure 8.2 A protective neuroinflammatory response. Injury to motoneuron cell bodies, or perhaps injury in the periphery, induces the release of repair signals from motoneurons, which promotes an M2 microglial protective phenotype. M2 microglia secrete increased levels of neurotrophic factors (NTFs), thereby exerting a neuroprotective function. M2 microglia also release anti-inflammatory cytokines to inhibit the production of pro-inflammatory responses. Astrocytes also participate in the neuroprotective process with secretion of NTFs and uptake of excess glutamate from synaptic clefts. Astrocytes also enhance the antioxidant capacity of neurons by releasing the glutathione precursor which is taken up by motoneurons for the synthesis of glutathione. The controversial role of T cells in ALS was resolved by the demonstration that their presence in mSOD1 mice plays an endogenous neuroprotective function by augmenting the protective potential of microglia and attenuating their toxic responses (Beers et al., 2008; Chiu et al., 2008). Another study using mSOD1 mice bred with different T cell–deficient mice reached a similar conclusion that T cells mediate a beneficial response; in both studies, T cell deficiency led to the attenuation of microglial protective responses and survival. Banerjee et al. (2008) also concluded that the passive transfer of ex vivo–activated CD4+ T cells into mSOD1 mice improved neurological function and life expectancy. More recently, a costimulatory pathway was demonstrated to be up-regulated in T cells in the blood of patients with ALS; modulating this costimulatory pathway in ALS mice delayed disease onset and reduced toxic microglial inflammatory responses (Lincecum et al., 2010). There was also controversy as to whether the proliferation and activation of microglia in ALS promoted survival or exacerbated neuronal death. Several groups have demonstrated that wild-type (WT) microglia or microglia expressing less mSOD1 promoted neuroprotection and extended survival of mSOD1 mice (Beers et al., 2006; Boillée et al., 2006). In vitro studies utilizing primary microglia–motoneuron co-cultures provided evidence that WT microglia were less neurotoxic than mSOD1 microglia due to their enhanced release of neurotrophic factors (NTFs) and attenuated release of free radicals and proinflammatory cytokines (Xiao et al., 2007). Marden et al. (2007) determined that dysregulated redox stress in ALS mice caused by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase subunits NOX1 and NOX2, expressed in microglia, significantly influenced the progression of motoneuron disease caused by mSOD1 expression; deletion of either Nox gene slowed disease progression and improved survival. Therefore, T cells and microglia, through their distinctive temporal and spatial contributions, can have both neuroprotective and cytotoxic functions depending on their different phenotypic activation states and the physiological conditions they encounter. SOD1 is a ubiquitously expressed cytosolic enzyme that converts highly reactive and toxic superoxide radicals (O2•−) to hydrogen peroxide (H2O2), which is subsequently converted to water and oxygen by a catalase. Mutations in the SOD1 gene are known to cause fALS. After the SOD1 mutations responsible for fALS were identified, researchers overexpressed these SOD1 mutations in transgenic mice and rats that subsequently developed a chronic progressive motoneuron disease. This disease reflected many clinical and pathological features of ALS including tremulousness, muscle weakness and atrophy, spasticity, progressive paralysis and ultimately death (Gurney et al., 1994). Similar overexpression of WT SOD1 did not induce an ALS-like phenotype, nor did a deletion of SOD1 gene, suggesting that the toxicity is not due to a loss or gain of SOD1 enzymatic activity but rather to a toxic gain of function. Genetic and chimeric mouse studies indicate that the mechanisms of motoneuron injury are ‘non-cell-autonomous’, a phrase that denotes that other cell types are involved in the pathoprogression of the disease. Mice chimeric for mSOD1 revealed that WT motoneurons, when surrounded by mSOD1-expressing glia, were injured, while mSOD1-expressing motoneurons surrounded by WT glia appeared normal (Clement et al., 2003). The lack or reduction of mSOD1 expression in microglia slowed disease progression and prolonged survival of mSOD1 mice, while disease onset was not altered. Reduced mSOD1 expression in astrocytes also slowed disease progression and prolonged survival, while not affecting disease onset (Yamanaka et al., 2008). The only cell type where mSOD1 expression was beneficial was the Schwann cell, where reduced expression of an enzymatically active mSOD1 in Schwann cells accelerated disease progression and reduced survival of mSOD1 mice (Lobsiger et al., 2009). While early attempts at overexpressing mSOD1 predominantly in neurons were unsuccessful at inducing motoneuron injury, high homozygous mSOD1G93A overexpression in mice did induce a motoneuron disease, although disease was dramatically delayed (Jaarsma et al., 2008). As in patients with ALS, neuroinflammation is a prominent pathological feature of mice expressing mSOD1, and again is characterized by activated microglia, infiltrating T cells, immunoglobulins and dendritic cells (DCs) in the spinal cords of these mice. Activated microglia are observed at an early age prior to signs of disease onset. Numerous studies of mSOD1 mice also suggest immune activation of microglia with increased expression of inducible nitric oxide synthase (iNOS), the catalytic subunit of the phagocyte NADPH oxidase (NOX2), interleukin-10 (IL10), IL6, tumour necrosis factor alpha (TNFα) and CCL2. Although it is not clear if these responses are protective or injurious, there is clear evidence of an early immune response in the transgenic mSOD1 mice. Combined, these results indicate that glial cells contribute significantly to ALS progression and that the disease is non-cell-autonomous, involving both motoneurons and surrounding glia. TDP43 is a multifunctional DNA- and RNA-binding protein involved in RNA processing, stabilization and transport. Its involvement in ALS pathogenesis was originally recognized when TDP43 was identified as a primary component of ubiquitinated inclusions in neurons and glial cells in sALS tissue, and it was then reinforced when TDP43 mutations were identified in fALS patients (Neumann et al., 2006). WT TDP43 and several TDP43 mutations have been overexpressed in neurons of mice and rats using the Prp, Thy1 or CaMKII promoters, which subsequently resulted in growth retardation, gait abnormalities, degeneration of motoneurons and pyramidal neurons in layer 5 of the frontal cortex, microglial and astroglial activation, cognitive impairments and premature death; the severity of the phenotype correlated with the levels at which the transgene accumulated in the neuron. In addition, abnormal nuclear inclusions composed of TDP43 and FUS–TLS are found in motoneurons, with substantial accumulations of mitochondria in cytoplasmic inclusions, a lack of mitochondria in axon terminals and immature neuromuscular junctions. TDP43 deletions are embryonically lethal, and a conditional deletion of a floxed TDP43 by a tamoxifen-induced Cre recombinase resulted in dramatic weight loss and death of the mouse in 9 days. Moderate overexpression of mTDP43 or truncated TDP43, as compared with equivalent expressions of WT TDP43, resulted in up-regulated cytoplasmic and nuclear ubiquitin and aggregates of phosphorylated TDP43 with enhanced motoneuron degeneration. Currently, less is known about the TDP43 rodent models than the SOD1 models due to their recent development. Different mechanisms of motoneuron injury and death in ALS have been proposed. These mechanisms include misfolded protein toxicity – particularly misfolded SOD1 or TDP43 toxicity; glutamate- and calcium-mediated excitotoxicity; mitochondrial dysfunction; neurofilament and cytoskeletal alterations affecting axonal transport; RNA processing and handling defects; endoplasmic reticulum (ER) and Golgi dysfunction, resulting in ER stress and protein degradation malfunction; neuroinflammation with subsequent neuroprotection and/or neurotoxicity; and astroglial alterations with reduced neuroprotection and direct neural toxicity. These mechanisms of neural injury have been proposed because all pathological alterations are present in the motoneuron and/or surrounding glia, and all contribute to neurodegeneration, although the initiating mechanism is not clear. In addition, these observed pathological processes interact with each other, aggravate one another and may even have a synergistic injurious effect. In this chapter, the currently proposed mechanisms of motoneuron protection and injury, induced by the accompanying neuroinflammation, will be discussed. Although the mechanisms whereby mSOD1 causes disease are unknown, there is compelling evidence suggesting immune system involvement in mSOD1-mediated motoneuron injury. Several studies have demonstrated the presence of activated microglia, T cells and DCs in the spinal cords of mice expressing the G93A form of mSOD1, with elevated transcripts in the mSOD1 spinal cord beginning at 110 days (Henkel et al., 2006). CCL2 mRNA and immunoreactivity were up-regulated in neuronal and glial cells as early as 15 days prior to any evidence of microglial activation. Zhong et al. (2008) also demonstrated an increase in CCL2 mRNA and suggested it was correlated with a breakdown in the blood–brain barrier. In addition, at 39 days of age, before evidence of disease onset, CD68 immunoreactivity, a myeloid-specific LAMP protein expressed by phagocytic cells, was present in lumbar spinal cords of mSOD1 mice. Other studies also indicate an immune activation in the mSOD1 mice, including activated microglia, increased iNOS expression, increased IL1β expression and increased IL6, TNFα and CCL2 expression. Although it is not clear if these responses were protective or injurious, there is clear evidence suggestive of an early immune response in the transgenic mSOD1 mouse model of fALS, as was shown in patients with ALS. As has been found in other models of neuronal injury, microglia activation may be a double-edged sword with the abilities to promote either neuronal protection or injury. In ALS patient autopsy tissues, the neuroinflammation is observed specifically at sites of motoneuron injury, highlighted by the presence of activated and proliferating microglia and infiltrating DC (Henkel et al., 2009; Philips and Robberecht, 2011). Positron emission tomography using [11C](R)-PK11195 enabled the detection of cortical microglial activation throughout disease in ALS patients (Turner et al., 2004). This microglial activation, which was not present in healthy controls, correlated with the severity of upper motoneuron damage. In addition, granular aggregates of misfolded SOD1 were regularly detected in ventral horn microglia in ALS autopsy tissues, regardless of whether the patients were carrying or lacking SOD1 mutations (Forsberg et al., 2011). ROS play a significant role in exacerbating disease in ALS by aggravating the pathological mechanisms of motoneuron injury. As evidence of damage, cerebrospinal fluid (CSF), serum and urine from ALS patients all contain elevated biomarkers of free radical damage. The reactive species involved in ALS include NO, O2•−, H2O2, hydroxyl radical (•OH) and peroxynitrite, the latter two being particularly toxic. O2•− is generated from two sources: the enzyme NADPH oxidase and mitochondrial respiration. About 15% of O2•− produced by mitochondria goes towards formation of peroxynitrite and the other 85% is converted to H2O2. NO is produced by the enzymes nNOS (expressed in neurons, and can be expressed in activated astrocytes), eNOS (expressed in endothelial cells) and iNOS (expressed in activated microglia); however, iNOS is also seen in motoneurons in early stages of disease in the mSOD1 mouse and in astrocytes as the disease progresses. While the direct toxicity of NO is modest, its toxicity is greatly enhanced by reacting with O2•−. NO readily diffuses into the mitochondria, where it rapidly reacts with O2•− to form the highly toxic peroxynitrite. NO can capture O2•− three times faster than SOD1, and in fact, NO is the only biological molecule produced in high enough concentrations to outcompete SOD1 for O2•−. NO and O2•−
Neuroimmunology of Amyotrophic Lateral Sclerosis
Overview of amyotrophic lateral sclerosis
Mutant superoxide dismutase animal model of ALS
TDP43 animal model of ALS
Proposed mechanisms of motoneuron injury in ALS
Neuroinflammation and mSOD1 mice
Immunologic aspects of ALS: part 1 – microglia
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel







Full access? Get Clinical Tree


