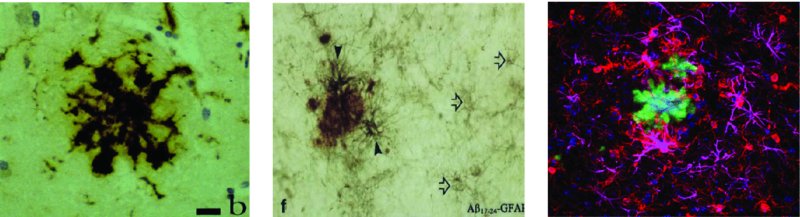
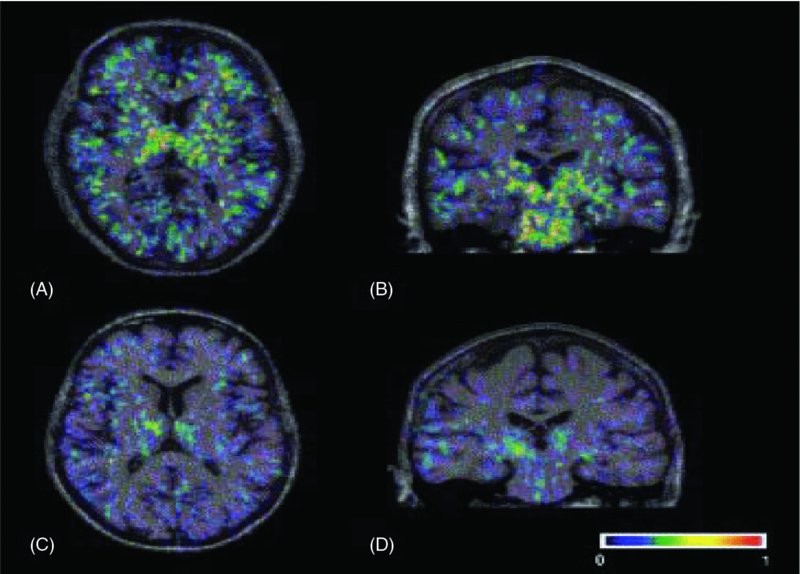
6 Magdalena Sastre1, Loukia Katsouri1, Amy Birch1, Alexander Renziehausen1, David T. Dexter1, Robert R. Crichton2 and Roberta J. Ward1,3 1Centre for Neuroinflammation and Neurodegeneration, Division of Brain Sciences, Imperial College London, London, UK 2Université Catholique de Louvain, Louvain-la-Neuve, Belgium 3Biologie du Comportement, Université Catholique de Louvain, Louvain-la-Neuve, Belgium Parkinson’s and Alzheimer’s disease are common neurodegenerative diseases which occur in individuals with advancing age (>65 years). Alzheimer’s disease (AD) initially presents with short-term memory loss, followed by widespread cognitive and motor impairment caused by a widespread neuronal loss in the hippocampus and selected cortical and subcortical regions. In Parkinson’s disease (PD), there is loss of motor co-ordination which is characterized by resting tremor, muscle rigidity, bradykinesia and postural instability. This is principally due to the loss of dopaminergic neurons in the substantia nigra (SN) which forms part of the basal ganglia which controls movement. Both AD and PD are polygenic diseases, meaning that a wide number of risk genes have been implicated in the development of these two neurodegenerative diseases of ageing. In contrast, Huntington’s disease (HD) occurs during midlife (at approximately 40–50 years of age), affects 1 in 10,000 individuals worldwide and is characterized by movement disorders, cognitive deterioration and psychiatric disturbances. The most sensitive region affected is the striatum, where there is loss of GABAergic medium-sized spiny striatal (caudate nucleus and putamen) neurons. HD is caused by an expansion of CAG repeats coding for a poly-Q tract in the huntingtin protein. In individuals with less than 35 CAG repeats, the likelihood of the disease developing is negligible. However, as the number of CAG repeats increases (i.e. to over 40 CAG repeats), the disease will develop. An autosomal dominant pattern of inheritance of this huntingtin gene has been demonstrated. The common feature of these three diseases is the presence of intracellular aggregates, or inclusion bodies containing misfolded proteins (i.e. amyloid-beta (Aβ), alpha-synuclein (contained within Lewy bodies) and huntingtin for AD, PD and HD, respectively). The exact roles of these aggregated proteins remain unclear; whether they are protective or are involved in the neuroinflammation of these diseases remains to be elucidated. Other proteins affected which may play a role include chaperones and elements of the cytosolic protein degradation pathway, such as ubiquitin and components of the proteosomal apparatus. The main pathological hallmarks of AD include the presence of neuritic plaques, neurofibrillary tangles, synaptic loss and, ultimately, neuronal death. Neuroinflammation in AD is characterized by an inflammatory response to Aβ, inducing the activation of microglia and the recruitment of astrocytes to the sites where Aβ deposits occur (Sastre et al., 2006b) (Figure 6.1). At present, it is still unclear whether inflammation is the cause or consequence of disease progression. However, there is evidence that suggests that it increases Aβ generation, tau phosphorylation and cognitive impairment. In this chapter, we will discuss all of these issues, including the cell types and molecular mediators involved. Figure 6.1 Glial cells surround amyloid-β deposits in human and transgenic mouse brain. (A) MHC class II antigen positive microglial cells display a circular distribution around the centre of the plaque in the brain of a 63-year-old human (Thal et al., 1997); and (B) fleecy amyloid is associated with a number of small GFAP-positive astrocytes in the human brain (Thal et al., 2000). Immunohistochemical detection for GFAP (magenta), IBA1 (red) and thioflavin-S (green) in the brain of an APP23 transgenic mouse. Aβ is able to bind and activate microglia. The mechanism of action for this is through interaction with pattern recognition receptors (PRRs). Microglia express many PRRs (Farina et al., 2007; Falsig et al., 2008) which recognize and bind PRR ligands, that is, pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs), such as Aβ (Salminen et al., 2009). Microglia interact with fibrillar Aβ through an ensemble of surface receptors composed of the alpha(6)beta(1) integrin, CD36, CD47 and the class A scavenger receptor (Reed-Geaghan et al., 2009). Interaction of microglia with Aβ via PRRs provokes their inflammatory actions (Solito and Sastre, 2012) (see Chapter 2). However, microglia internalize soluble Aβ from the extracellular milieu through a nonsaturable, fluid-phase macropinocytic mechanism that is distinct from phagocytosis and receptor-mediated endocytosis both in vitro and in vivo (Mandrekar et al., 2009). Clinical reports and studies in animal models suggest that microglial activation precedes amyloid plaque and tangle formation (Griffin et al., 1989; Heneka et al., 2005), while positron emission tomography (PET) studies have reported inflammatory changes in one-third of amnestic mildly cognitively impaired subjects (Cagnin et al., 2001; Okello et al., 2009). Two-photon microscopy has clarified the anatomic relationships of microglia to Aβ, showing that microglia sample and react to Aβ deposits in transgenic mouse models. In this report, recruitment of microglia to newly formed plaques occurred within a few days (Meyer-Luehmann et al., 2008), while other investigations revealed the establishment of a dynamic interface between microglial processes and Aβ deposits (Bolmont et al., 2008; Koenigsknecht-Talboo et al., 2008). These studies do not fully agree with previous reports (Heneka et al., 2005), a finding that may depend on the animal model used. It was proposed that early microglial activation in AD delays disease progression by promoting clearance of Aβ before the formation of senile plaques, suggesting that glial activation is protective early in the disease (Wyss-Coray et al., 2003; Maragakis and Rothstein, 2006; Wyss-Coray, 2006). Studies have shown that bone marrow–derived macrophages (BMDMs) are able to efficiently eliminate amyloid and confer neuroprotection by secretion of growth factors such as the glia-derived neurotrophic factor (GDNF) which are potentially beneficial to the survival of neurons (Liu and Hong, 2003). Activated microglia in early stages of AD can reduce Aβ accumulation by increasing its phagocytosis, clearance and degradation (Frautschy et al., 1998; Qiu et al., 1998). The mechanism by which Aβ is phagocytosed depends on the physical properties of Aβ and whether it is soluble or fibrillar. Secreted Aβ1–40 and Aβ1–42 peptides are constitutively degraded by neprilysin and the insulin-degrading enzyme (IDE), a metalloprotease released by microglia and other neural cells, whose enzymatic activity is enhanced by inflammatory events, such as lipopolysaccharide (LPS) stimulation (Qiu et al., 1997). In later stages, with persistent production of pro-inflammatory cytokines, microglia lose their protective effect (Hickman et al., 2008; Jimenez et al., 2008) and may become detrimental through the release of cytokines and chemokines (Hickman et al., 2008). These inflammatory mediators modulate immune and inflammatory function and may also alter neuronal function. In addition, microglia from aged transgenic APP–PS1 mice have a decrease in the expression of the Aβ-binding scavenger receptors A (SRA), CD36 and RAGE and the Aβ-degrading enzymes IDE, neprilysin and matrix metalloproteinase-9 (MMP9), compared with wild-type controls (Hickman et al., 2008). Therefore, evidence supports the idea that over-activated microglia could cause uncontrolled inflammation that may drive the chronic progression of AD by exacerbating Aβ deposition and stimulating neuronal death (Mrak and Griffin, 2005; Gao and Hong, 2008). This concept constitutes the ‘neuroinflammatory hypothesis’. By comparison, the ‘microglial dysfunction hypothesis’ stipulates that rather than an increase of inflammatory function, there is a loss of the microglial neuroprotective function in AD (Polazzi et al., 2010). Research has shown that the phagocytic abilities of microglia are altered in aging and impaired in neurodegenerative diseases. Therefore, this ‘senescent’ or dystrophic microglia can also contribute to the onset of sporadic AD (Streit et al., 2004, 2009). The actual role of astrocytes in AD still remains elusive, because astrocytes seem to adopt different functions depending on disease progression and the extent of the accompanying parenchymal inflammation. When healthy astrocytes are exposed to high levels of certain pro-inflammatory cytokines, reactive oxygen species or even Aβ, they undergo a process called astrogliosis whereby they become activated. Astrogliosis is a defensive transformation in response to inflammation during which astrocytes undergo a complex morphological and functional remodelling involving hypertrophy, altered gene expression and up-regulation of inflammatory mediators (Sofroniew, 2009). If the inflammatory response is severe or particularly persistent, as it is in AD, reactive astrocytes will also proliferate to form a glial scar in an effort to isolate damaged tissue (Rodríguez et al., 2009; Medeiros and LaFerla, 2013). While there is no significant correlation between plaque load and cognitive deterioration, studies on brain tissues obtained from aged AD patients show a correlation between astrogliosis and cognitive decline, which suggests that astrogliosis may be partially responsible for the synaptic dysfunction that is undoubtedly implicated in the deterioration of brain function during AD (Brenner, 2005; Simpson et al., 2010; Verkhratsky et al., 2010). Although the exact processes underlying this dysfunction remain elusive, it is well established that astrogliosis leads to elevated expression and secretion of cytokines and chemokines, whose accumulation results in neurotoxicity (Li et al., 2011). Reactive astrocytes also up-regulate inducible nitric oxide synthase (iNOS), stimulating not only high concentrations of astrocytic neurotoxic nitric oxide (NO) but also nitration of the Aβ tyrosine at position 10, which can potentiate the propensity of this amyloidogenic protein to self-aggregate (Kummer et al., 2011). In addition, astrocytes also contribute to neuronal death through release of glutamate. However, it has also been suggested that reactive astrocytes play a beneficial role during AD, for example by being directly involved in the clearance of Aβ by both phagocytosis and secretion of Aβ-degrading proteases such as neprilysin, as well as apolipoprotein E (ApoE) (Apelt et al., 2003; Wyss-Coray et al., 2003; Koistinaho et al., 2004; Pihlaja et al., 2007, 2011; Terwel et al., 2011). Reactive astrocytes are essential in limiting inflammation and neuronal damage in models of acute central nervous system (CNS) injury (Faulkner, 2004; Myer, 2006). Therefore, it is plausible that in chronic CNS diseases, such as AD, reactive astrocytes may initially play a similar protective role while prolonged activation gradually reduces their capacity to act in a neuroprotective manner. Interestingly, preventing astrocyte activation at an early stage of pathology (through gene deletion of GFAP and vimentin in an AD mouse model) results in an increased plaque load accompanied by neuronal dystrophy. These studies underscore the idea that reactive astrocytes could indeed play a neuroprotective role during early disease stages, which may gradually be outweighed by increased cytokine production, oxidative stress and excessive Aβ production exhibited by reactive astrocytes with disease progression. Several studies have attempted to determine at what stage of AD astrogliosis occurs and how it could impact clinical disease. Reactive astrocytes become plaque associated at early stages in both transgenic mice and AD patients (Kato et al., 1998; Heneka et al., 2005; Van Dam et al., 2005; Carter et al., 2012). To track the extent of astrogliosis with the progression of amyloid pathology, an animal model of AD was used where transgenic APP23 mice were crossed with glial fibrillary acidic protein (GFAP)-luc mice, which express luciferase under the control of the GFAP promoter (Watts et al., 2011). This study showed that in vivo bioluminescence imaging levels increased at 14 months of age, which correlates to a late activation of astrocytes when AD pathology had already progressed considerably. Together the studies presented here suggest that astrocytes have evolved to deal with acute localized inflammation, rather than chronic disseminated inflammation, and that astrogliosis represents a common mechanism by which the brain attempts to deal with early disruptions caused by trauma or neurodegenerative disease. Aβ can increase gene expression and protein production of a number of cytokines by human microglia in vitro, including interleukin-1 beta (IL1β), IL8, IL10, IL12, tumour necrosis factor alpha (TNFα), macrophage inflammatory protein-1 alpha (MIP1α, aka CCL3), MIP1β (CCL4) and monocyte chemoattractant protein-1 (MCP1, CCL2) (Lee et al., 2002). It is no surprise, therefore, that their expression is reported to be altered in the brain, blood and cerebrospinal fluid (CSF) of AD patients (Akiyama et al., 2000). Animal models of AD, such as the APP transgenic line Tg2567 carrying the Swedish mutation, also show enhanced levels of TNFα, IL1β, IL1α, chemoattractant protein-1, COX2 and complement component 1q (Sastre et al., 2008). Despite the strong links between inflammation and AD, there are a surprisingly small number of studies assessing the expression of many inflammatory cytokines. Evidence of an increased risk of AD is observed when the IL10−1082 GA gene polymorphism is present; however, the association is only marginally significant (Di Bona et al., 2012). Adeno-associated virus (AAV)-mediated gene delivery of this anti-inflammatory cytokine reduced glial activation, enhanced Aβ clearance into the plasma, upregulated neurogenesis and improved spatial memory in APP–PS1 mice (Kiyota et al., 2012); however, it had no effect on Aβ plaque load. Increased concentrations of p40 (a common subunit of the pro-inflammatory cytokines IL12 and IL23) are found in the CSF of AD patients, and this correlates with cognitive performance (vom Berg et al., 2012). In addition, ablation of IL12 and IL23 signalling in APP–PS1 mice reduces amyloid burden (vom Berg et al., 2012). IL6 is elevated in the serum of patients with AD (Helmy et al., 2012), and there is an interaction between IL6 and IL10 gene polymorphisms associated with increased risk for AD (Arosio et al., 2004; Combarros et al., 2009). These studies strongly suggest that cytokine dysregulation is a significant candidate for initiating the development of AD. The most commonly studied cytokines are IL1β and TNFα; up-regulation of these pro-inflammatory cytokines has been heavily implicated as common cellular response markers to Aβ release and deposition (Hickman et al., 2008). IL1 is a primary mediator of the pro-inflammatory response in the body and is capable of inducing the production of many other cytokines. A number of studies have analysed the possibility that polymorphisms in IL1 could lead to increased risk for the development of AD. A specific IL1α gene polymorphism in allele 2 triples the risk of developing AD (Du et al., 2000; Grimaldi et al., 2000; Rebeck, 2000), and an even greater risk is seen with patients carrying both IL1α and IL1β allele 2 gene polymorphisms (Griffin et al., 2000). In addition, this IL1α gene polymorphism is associated with increased microglial cell numbers in post-mortem AD brains, and the increase was even higher in patients carrying the APOE ϵ4 allele (Hayes et al., 2004). These polymorphisms increase expression of IL1 and may therefore drive IL1-mediated mechanisms that lead to an overall enhancement of neuroinflammation in the brain and consequently increased risk for AD. Overexpression of IL1β has been reported in microglia and astrocytes of AD brains since the late 1980s (Griffin et al., 1989), in particular in plaque-associated microglia (Mrak and Griffin, 2005). This association is transient; high IL1β expression is associated with early amyloid deposits and the conversion of diffuse amyloid plaques into compact forms (Griffin et al., 1995). IL1α and IL1β increase translation of APP in astrocytes in vitro, with the latter enhancing synthesis to a greater extent (Rogers et al., 1999). In addition, IL1β drives astrocyte activation and increases β-secretase-cleaved carboxyl-terminal fragment (βCTF) production in vivo, suggesting that this cytokine may have a significant role to play in the progression of AD (Sheng et al., 1996). Indeed, many argue that early and sustained overexpression of IL1 could be an initiator of disease pathology (Griffin and Mrak, 2002). IL1 may also contribute directly to neuronal dysfunction in AD; chronic release of exogenous IL1β in vivo induced overexpression and phosphorylation of neurofilament protein and tau, which are components of neurofibrillary tangles of AD (Sheng et al., 2000). This overexpression was specifically associated with swollen, dystrophic neurites. Interestingly, a recent paper by Ghosh and colleagues reports that sustained IL1β overexpression in a triple transgenic (3xTg) mouse model of AD increases tau hyperphosphorylation but leads to a corresponding decrease in amyloid burden (Ghosh et al., 2013). They also found a four- to sixfold increase in plaque-associated microglia, suggesting that IL1β may directly affect tau phosphorylation, possibly via p38 mitogen-activated kinase and glycogen synthase kinase-3β activity, but regulates amyloid indirectly via glial activation. This builds on similar findings from their group showing that overexpression of IL1β specific to the hippocampus in amyloid precursor protein–presenilin-1 (APP–PS1) mice induces significant astrocyte and glial activation with a corresponding decrease in amyloid plaque deposition (Shaftel et al., 2007). The differential effect of IL1 on tau and amyloid makes it an unlikely candidate for therapeutic manipulation; however, it does lead to intriguing questions about targeting certain microglial or astrocytic processes to reduce amyloid burden. TNFα is increased in the serum of AD patients (Fillit et al., 1991; Bruunsgaard et al., 1999; Tarkowski et al., 1999), and increased expression is found in activated microglia surrounding amyloid plaques in brains of AD patients (Dickson et al., 1993). Microglia produce TNFα that is neurotoxic in response to Aβ exposure (Meda et al., 1995; Combs et al., 2001), and Aβ itself can induce neuronal apoptosis via TNF receptor type I (TNF-RI) (Li et al., 2004). These studies indicate that there may be TNFα-mediated apoptosis in AD. Increased TNFα is also found in 3xTg-AD and APPswe mouse models, expressed by both neurons and microglia (Mehlhorn et al., 2000; Janelsins et al., 2005). Overexpression of TNFα in 3xTg-AD mice induces significant neurodegeneration (Janelsins et al., 2008), providing further evidence for its role in the cell death seen in AD. However, chronic down-regulation of TNF-RII in 3xTg-AD mice exacerbates amyloid and tau pathology (Montgomery et al., 2013), although interestingly this effect was evident only in mice whose TNF-RII receptors were knocked down from 12 to 21 months of age. Young and aged mice in which TNF-RI or both TNF-RI and TNF-RII receptors were knocked down showed no change in amyloid burden or tau hyperphosphorylation. TNF-RI and TNF-RII show divergent signalling pathways, with TNF-RI being heavily implicated in neuronal apoptosis (Yang et al., 2002), whereas TNF-RII is reported to activate alternative nuclear factor kappa B (NF-κB) signalling associated with the down-regulation of pro-inflammatory cytokines (Rauert et al., 2010). This once again highlights the complex nature of cytokine involvement in AD, and any therapeutic intervention targeted at TNFα must take into account disease state and receptor specificity. Pro-inflammatory cytokines are known to affect Aβ generation, suggesting that a positive feedback loop may develop following an Aβ-triggered inflammatory response in which cytokines contribute to further Aβ production (Sastre et al., 2006a,b). The potential mechanisms include increasing the susceptibility for Aβ deposition or aggregation (Games et al., 1995; Guo et al., 2002), up-regulating β-secretase (BACE1) mRNA (Sastre et al., 2003, 2006b) and elevating APP transcription (Amara et al., 1999; Rogers et al., 1999). ROS are chemically reactive molecules that are cytotoxic by-products of oxygen metabolism. These include hydroxyl radicals (−OH), peroxide (H2O2) and superoxide radicals (O2−). Endogenous ROS are mainly produced by mitochondria, peroxisomes, endoplasmic reticulum and the NADPH oxidative complex (NOX) in the cell membranes (Vitale et al., 2013). The main mechanisms for eliminating ROS are superoxide dismutases (SODs), catalase, heme-oxygenase and glutathione peroxidase, enzymes that are increased in the vicinity of Aβ plaques (Furuta et al., 1995; Pappolla et al., 1998). During ageing or inflammation, there is accumulation of ROS, which causes oxidation of lipids, sugars, proteins and DNA, thus leading to cell dysfunction and consequently cell death (Chami and Checler, 2012). The neurons are highly susceptible to oxidative stress because of their high metabolic demands and poor anti-oxidant capabilities. ROS can also contribute to further inflammation through induction of cyclooxygenase-2 (COX2), inflammatory cytokines and pro-inflammatory transcription factors such as NF-κβ (Yan et al., 1995). In AD brains, several oxidative markers are increased (Guglielmotto et al., 2010), including protein oxidation (Smith et al., 1997), lipid peroxidation (Mark et al., 1997; Sayre et al., 1997), advanced glycation end products (Smith et al., 1994) and oxidation of nucleic acids (Nunomura et al., 1999). Oxidative stress is greatest early in the disease (Nunomura, 2001). Increased levels of isoprostanes, a marker for lipid peroxidation, were detected in the urine, blood and CSF of both mild cognitive impairment (MCI) and AD patients (Praticò et al., 2000, 2002; Mecocci, 2004), suggesting that oxidative markers could be potentially used as biomarkers for the early detection of AD. Increased ROS and oxidative stress were also confirmed in mouse models of AD (Pappolla et al., 1998; Smith et al., 1998; Matsuoka et al., 2001; Praticò et al., 2001). It is well established that Aβ causes oxidative stress which in turn causes more Aβ, resulting in a positive feedback loop that further contributes to the progress of the disease (Tamagno et al., 2012; Butterfield et al., 2013). This is because ROS affect the rate-limiting enzyme of AD, beta-site APP cleaving enzyme-1 (BACE1), by altering its distribution in non-apoptotic conditions (Tan et al., 2013) as well as its activity and expression (Tamagno et al., 2002). Oxidative stress-mediated BACE activity is γ-secretase-dependent as absence of PS1 abolished the effect (Tamagno et al., 2008). Further studies strengthened the correlation of BACE1, γ-secretase and oxidative stress, showing that treatment with dehydroepiandrosterone, an antioxidant, reduced BACE1 activity in NT2 neurons exposed to oxidative stress (Tamagno et al., 2003) and that mild impairment of oxidative metabolism by thiamine deficiency increased Aβ, carboxyl-terminal fragments and plaque burden in an AD mouse model (Karuppagounder et al., 2009). Dysfunctional homeostasis of transition metals, such as copper and particularly iron (Bush, 2013), may play an important role in the pathogenesis of AD by enhancing oxidative stress. (Crichton et al. 2011). Increased concentrations of iron are present in the basal ganglia (Bartzokis and Tishler, 2000), hippocampus (Ding et al., 2009) and neocortex (Cornett et al., 1998) and in or around the plaques and tangles (Morris et al., 1994). In addition, iron modulates the ability of α-secretase to cleave APP (Rivera-Mancía et al., 2010), promotes Aβ toxicity (Bodovitz et al., 1995) and aggregation, and directly regulates the expression and the synthesis of APP via the iron responsive element at the 5′ untranslated region of APP mRNA (Rogers et al., 2002; Rivera-Mancía et al., 2010). Levels of furin, an enzyme responsible for the activation of β-secretases involved in the amyloidogenic pathway, are also modulated by iron (Crichton et al., 2011), which will play an important role in AD pathogenesis (Silvestri and Camaschella, 2008). Complement receptors are one of the categories of cell surface molecules on microglia that are up-regulated in response to the activation of these cells (Liu and Hong, 2003). Aβ-induced complement activation leads to generation of C1q, C4 and C3 activation fragments around the plaques. Here microglia express complement proteins C1q and C3 and receptors C1qR, CR3, CR4 and C5aR, which support phagocytic uptake (Keene et al., 2011). Inhibition of the complement system results in an increase of Aβ plaque formation and neurodegeneration in AD transgenic mice (Shen and Meri, 2003). In contrast, lack of C1q in mouse models of AD results in decreased pathology (Hafer-Macko et al., 2000). This indicates that one mechanism by which microglia could recruit further reactive cells to the site of a plaque and cause neurotoxic damage is by activating the classical complement pathway and the inflammatory machinery associated with it (i.e. pro-inflammatory cytokines and oxidative products) through production of C1q (McGeer and McGeer, 1998; Bonifati and Kishore, 2007). There have been a number of reports demonstrating the presence of T cells in the brain of AD patients since the original observation 25 years ago (Rogers et al., 1988). Furthermore, inflammatory IFNγ-secreting Th1 cells and IL17–secreting Th17 cells have been shown to infiltrate the brain of older APP–PS1 mice (Browne et al., 2013). Therapeutic vaccination with Aβ antibodies in mice evidenced the Fc-mediated uptake and clearance of Aβ antibody complexes by local activated microglia (Bard et al., 2000). However, because a human trial of Aβ immunization led to meningoencephalitis in some patients, this treatment has been discontinued. Recently, it was found that nasal vaccination in mice was able to decrease Aβ, and the extent of this reduction correlated with microglial activation, suggesting that this may be a promising approach for human Aβ immunization (Frenkel et al., 2005). The finding that treatment with non-steroidal anti-inflammatory drugs (NSAIDs) is associated with a reduced risk and age of onset of AD reinforced the hypothesis that modulating inflammation could have therapeutic efficacy. The beneficial effects of NSAIDs have also been associated with reductions in Aβ generation, because experiments in vitro and in AD animal models indicate that certain NSAIDs are able to decrease Aβ levels, plaque size and tau phosphorylation (Yoshiyama et al., 2007; El Khoury and Luster, 2008; Sastre and Gentleman, 2010). However, clinical trials have failed to reproduce the beneficial effects of NSAIDs in AD patients. This has led to further analysis of the previously published epidemiological data, which has revealed that the use of NSAIDs prevents cognitive decline in older adults if started in midlife (prior to age 65) rather than late in life (Hayden et al., 2007). In addition, it was recently shown that while NSAIDs may indeed protect those with healthier brains, they can accelerate AD pathogenesis in patients with advanced stages of the disease (Breitner et al., 2009). This is supported by studies in transgenic mice, in which NSAIDs can prevent the appearance of cell cycle protein markers in neurons in young mice, but not after cell cycle entry has been initiated (Varvel et al., 2009). Therefore, it seems that the protective effects of NSAIDs depend very much on the stage of the disease at which the medication is started as well as the duration of the treatment (Sastre and Gentleman, 2010). PPARγ inhibition regulates the transcription of pro-inflammatory genes, such as IL1β; therefore, activation of PPARγ consequently inhibits the inflammatory response. In addition, our group found that PPARγ activators are able to decrease total Aβ levels under inflammatory conditions by affecting BACE1 transcription (Sastre et al., 2003, 2006b). Recently it was shown that ibuprofen is able to suppress RhoA activity in neuronal cells through PPARγ activation, promoting neurite elongation (Dill et al., 2010). Therefore, PPARγ activation could be beneficial in AD at several levels. However, other groups have suggested that PPARγ may affect Aβ clearance and degradation. Landreth demonstrated that PPARγ activation induces lxrα, apoe, and abca1 expression, promoting Aβ clearance by both microglia and astrocytes (Mandrekar-Colucci et al., 2012). Furthermore, PPARγ stimulates an increase of Aβ phagocytosis, which was mediated by the up-regulation of scavenger receptor CD36 expression. In addition, combined treatment with agonists for the heterodimeric binding partners of PPARγ, the retinoid X receptors (RXRs), showed additive enhancement of the Aβ uptake that was mediated by RXRα activation (Yamanaka et al., 2012). A recent prospective randomized, open-controlled study with pioglitazone (a typical PPARγ agonist) showed that at 6 months, the Wechsler Memory Scale-Revised logical memory-I scores significantly increased in the pioglitazone group, but not in the control group (Hanyu et al., 2009). Another PPARγ agonist, rosiglitazone, has been trialled with inconsistent results. In contrast to pioglitazone, rosiglitazone cannot cross the blood–brain barrier (BBB) (Festuccia et al., 2008), and it was suggested that the protective effects are mediated through its effects on insulin and glucocorticoids that are able to penetrate the brain. Minocycline, a tetracycline derivative, has potent anti-inflammatory, anti-apoptotic and neuroprotective properties. Minocycline is able to cross the BBB and effectively delays disease progression and reduces neuronal death in mouse models of amyotrophic lateral sclerosis (Zhu et al., 2002), Huntington’s disease (Chen et al., 2000) and Parkinson’s disease (Wu et al., 2002). In many cases, the neuroprotective properties of minocycline have been attributed to inhibition of caspases. In primary cortical neurons, minocycline was shown to reduce caspase-3 activation and lowered the generation of caspase-3-cleaved tau fragments (Noble et al., 2009). Recently, minocycline was shown to protect against Aβ-induced cell death and prevent fibrillization of Aβ in vitro (Familian et al., 2006), reduce iNOS levels (Ferretti et al., 2012), prevent Aβ deposition and cognitive decline in APP transgenic mice (Seabrook et al., 2006; Ferretti et al., 2012) by reducing BACE1 levels (Ferretti et al., 2012) and inhibit neuronal death and attenuate learning and memory deficits following administration of Aβ to rats (Ryu et al., 2004; Choi et al., 2007). In addition, treatment of tangle-forming transgenic mice (htau line) with minocycline resulted in reduced levels of tau phosphorylation and insoluble tau aggregates (Noble et al., 2009). Another potential mechanism of action of minocycline has been related to the inhibition of microglial activation. Administration of minocycline in animal models of ALS attenuated the induction of the expression of M1 microglia markers during the progressive phase, whereas it did not affect the transient enhancement of expression of M2 microglia markers during the early pathogenesis phase (Kobayashi et al., 2013). This study suggests that minocycline may selectively inhibit the microglia polarization to a pro-inflammatory state. Interestingly, anti-TNFα treatment reduced Aβ and tau phosphorylation in transgenic mice. Treatment with the antibody against TNFα, infliximab, increased the number of CD11c-positive dendritic-like cells and the expression of CD11c. These data suggested that the CD11c-positive dendritic-like cells might contribute to the infliximab-induced reduction of AD-like pathology (Shi et al., 2011). Although the disease mechanisms involved in PD are not fully understood, the progressive neurodegeneration of dopaminergic neurons in the SN that occurs in PD over a number of years is associated with chronic inflammation. This is driven by the inflammatory response of microglia cells, reactive microgliosis. Early evidence for the involvement of neuroinflammation in PD was reported by McGeer and colleagues (1988b), who identified reactive microglia in the SN of post-mortem brains of PD patients +/− dementia, by immunohistochemical techniques, using HLA-DR-specific antibodies. Further immunohistochemical studies by Imamura et al. (2003) similarly identified major histocompatibility complex class II (MHC-II)-positive microglia, not only in the SN but also in the hippocampus, transentorhinal cortex, cingulate cortex and temporal cortex in PD brains with a range of 4–14 years of disease duration. Furthermore, the numbers of activated microglia increased in the SN and putamen as the neuronal degeneration of the SN proceeded. In contrast, Mirza et al. (2000) showed activation of microglia only in the SN of PD autopsies verified by immunohistochemical detection of MHC-II and ferritin. Overall clinical and animal studies have supported the role for activated microglia in PD (reviewed by Collins, 2012). In in vivo studies, where PET was utilized in PD patients, activated microglia, which were identified with the translocator protein (TSPO) ligand PK11195, [1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinoline carboxamide], the ‘peripheral benzodiazepine-binding site’ on activated microglia, showed increased binding potential, values only evident in the midbrain of PD patients in early disease stages (Ouchi et al., 2005) (see Figure 6.2). In PD patients with more advanced disease, there were significant increases in mean [11C](R)-PK11195 binding in the subcortical region, which included the striatum, pallidum, thalamus and pons (Gerhard et al.,2006). Such PET results indicated that there might be different patterns of microglia activation between early- and late-stage PD patients. In addition, these results indicated that microglia become activated very early in the disease process and remained activated for a long duration. Figure 6.2 Transverse and coronal sections of binding potential maps co-registered to the individual MRI. In the PD patient (A and B), binding is increased in the basal ganglia, pons and frontal regions, while the healthy control person (C and D) only shows constitutive [11C](R)-PK11195 binding in the thalamus and pons. (From Gerhard, A., Pavese, N., Hotton G., et al., 2006. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiology of Disease 21, 404–412, Elsevier.) Only a certain neuronal cell population (i.e. the dopaminergic neurons in the SN) are affected in PD, which is caused by the pro-oxidative state in which these neurons function relative to other brain regions; that is, they have an increased metabolic rate and a high content of (i) oxidizable products, such as dopamine; (ii) iron; and (iii) polyunsaturated fatty acid, but also, most importantly, a low content of antioxidants such as reduced glutathione (GSH) (reviewed by Niranjan, 2013). It seems that once the dopaminergic neuron destruction has begun, a self-renewing cycle of microglia activation (i.e. microgliosis) ensues. The vulnerability of these dopaminergic neurons was demonstrated with in vitro studies of mixed mesencephalic neuron–glia cultures where the soluble neuron factor, mu-Calpain, was released from damaged dopaminergic neurons, which activated microglia via activation of NADPH oxidase (Levesque et al., 2010). Such activated microglia in the nigrostriatal dopamine region will release prostaglandins, chemokines, enzymes associated with inflammation (e.g. iNOS and COX2) as well as pro-inflammatory cytokines (e.g. TNFα, IL1β, IL2, IL4, IL6, transforming growth factor alpha (TGFα), TGFβ1 and TGFβ2) (Dexter, 2013). Decreased secretions of brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) by the microglia will further compromise the neurons. In addition, the death-signalling receptor TNFR1 is expressed on dopaminergic neurons in human SN, which will contribute to their demise; blockade of this receptor in animal studies attenuated the death of dopaminergic neurons (reviewed in Collins, 2012). The phagocytosis of extracellular aggregated α-synuclein will further activate microglia and propel dopaminergic degeneration (Zhang et al., 2005). In vivo and in vitro studies where extracellular α-synuclein was either incubated with BV2 microglia cells or intra-nigrally injected into mouse brains, respectively, resulted in a positive NF-κB response with the production of pro-inflammatory cytokines (Couch et al., 2011). Furthermore, it has been suggested that the α-synuclein had a ‘priming effect’ on the microglia which will make them more susceptible to subsequent challenges from direct environmental pro-inflammatory challenge or via peripheral systemic inflammation (Morgan and Liu, 2011). In addition to microglial activators, there may also be changes in several anti-inflammatory systems in PD that play a role in regulating microglia activation. These include the CD200–CD200R receptor, the vitamin D receptor, PPARs and the soluble receptor for advanced glycation end products (reviewed by Ward et al., 2011). Astrocytes, the most numerous of the glial cells, are a major class of cells in the mammalian brain, outnumbering neurons by several-fold (Sherwood et al., 2006). Astrocytes are present in all brain regions and in close contact with neuronal structures, playing a critical and integral role in mediating the physiologic and pathologic states of neurons and the integrity of the BBB. Astrocytes can release and supply various neurotrophic factors (e.g. NGF, neurotrophin-3), metabolic substrates (e.g. lactate) and reduced GSH to ensure the survival and correct functioning of the neurons. It is of note that the concentration of GSH in astrocytes (approximately 3.8 mM) is estimated to be higher than that of neurons (approximately 2.5 mM) (Rice and Russo-Menna, 1998), probably as a result of higher specific activity of the γ-glutamylcysteine synthetase in astrocytes (Gegg et al., 2003). Uniquely astrocytes can release this antioxidant into the extracellular space to adjacent neurons via the transporter multidrug resistance protein, where it is cleaved by γ-glutamyltranspeptidase on the astrocytic plasma membrane, to generate precursors for neuronal GSH synthesis. The GSH content in the SN of PD patients has been observed to be significantly reduced (40%) (Sian et al., 1994), although the explanation for this loss remains unclear (reviewed by Rappold and Tieu, 2010). In addition, astrocytes are positioned close to synaptic clefts to remove excess glutamate, as well as potassium and calcium. However, if activated in response to neuronal damage or a toxic insult, cytokines and chemokines will be released which can have an adverse effect on neurons (reviewed by Rappold and Tieu, 2010). Although Mirza and colleagues (2000) could not identify reactive astrocytosis in SN and putamen of post-mortem tissues from PD patients, later studies revealed an active astrogliosis (reviewed by Niranjan, 2013) that could maintain the degeneration of the dopaminergic neurons. In support of this, astrocytic α-synuclein-immunoreactive inclusions have recently been noted to develop in sporadic PD. α-synuclein-labelled astrocytes occur preferentially in prosencephalic regions (the amygdala, thalamus, septum, striatum, claustrum and cerebral cortex). The topographical distribution pattern of these astrocytes closely parallels that of the cortical intraneuronal Lewy neurites and Lewy bodies (Braak et al., 2007). Mice injected with the toxin (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) (MPTP) and then killed at different time points showed a direct correlation between reactive astrocyte formation and dopaminergic structure destruction in both striatum and SN. Interestingly, although the neuronal death ultimately declined, GFAP) expression remained up-regulated (reviewed by Niranjan, 2013). Other brain regions (e.g. the frontal cortex and caudate nucleus) also showed an increase in expression of GFAP in PD brains, although the authors concluded that these astrocytes protected non-SN brain regions from oxidative and mitochondrial damage (Mythri et al.
Neuroinflammation in Alzheimer’s, Parkinson’s and Huntington’s Diseases
General introduction
Alzheimer’s disease
Microglia in AD
Astrocytes in AD
Molecular mediators in AD
Cytokines
Reactive oxygen species (ROS) in AD
Complement in AD
Adaptive immunity in AD
Anti-inflammatory therapy in AD
Non-steroidal anti-inflammatory drugs (NSAIDs)
Peroxisome Proliferator-activated Receptor Gamma (PPARγ) agonists
Minocycline
Anti-TNFα
Neuroinflammation in Parkinson’s disease
Microglia in PD
Astrocytes in PD
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel







Full access? Get Clinical Tree


