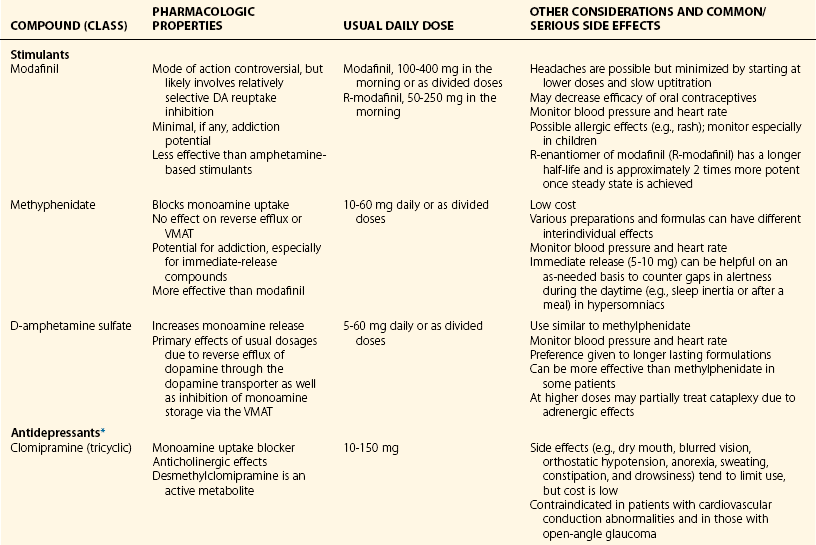
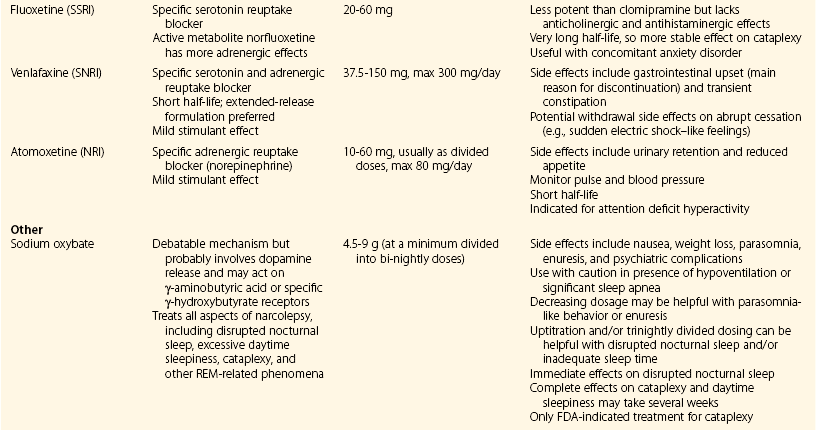
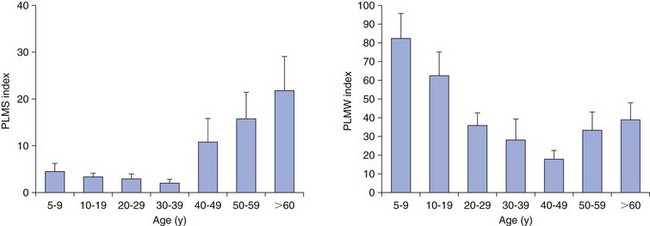
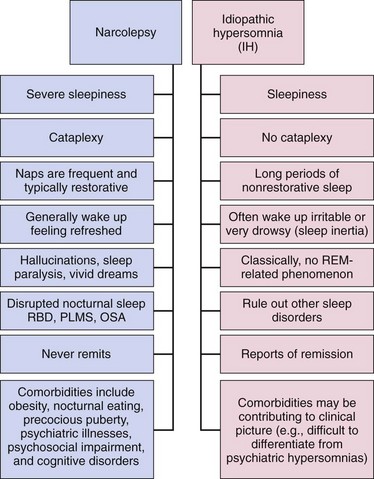
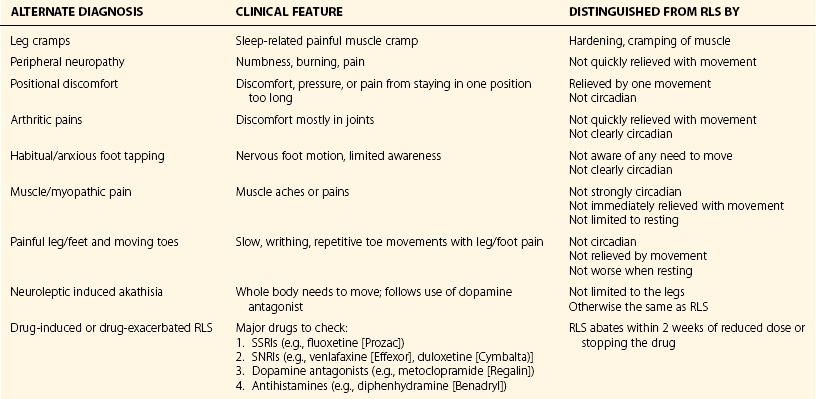
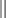Chapter 11 11.1 Central Nervous System Hypersomnias The classification of CNS hypersomnias is being revised for the International Classification of Sleep Disorders, third edition (ICSD-3). The ICSD-3 will further subdivide narcolepsy with and without cataplexy into narcolepsy with and without hypocretin deficiency. The terms type 1 narcolepsy and type 2 narcolepsy may be introduced to denote the presence or absence of hypocretin deficiency, respectively, to parallel the accepted nomenclature for diabetes. For the purpose of this chapter, we refer to the current (second) International Classification of Sleep Disorders (ICSD-2), summarized in Box 11.1-1. CNS hypersomnias may manifest as a variety of symptoms, including cataplexy, sleep paralysis, hypnagogic and hypnopompic hallucinations, EDS, disrupted nocturnal sleep, automatic behaviors, and sleep inertia. Narcolepsy typically manifests more of these symptoms than idiopathic hypersomnia (IH) does, but these symptoms, excluding cataplexy, may also occur outside the context of a CNS hypersomnia in the general population (Fig. 11.1-1). Figure 11.1-1 Comparison of narcolepsy and idiopathic hypersomnia. Except for cataplexy, which is pathognomonic for narcolepsy with hypocretin deficiency, no symptoms are unique to CNS hypersomnias. Typical cataplexy is described as the sudden onset of a transient loss of skeletal muscle tone that may last from seconds to minutes, most often triggered by positive emotions, such as laughter and joking. Cataplexy occurs in 60% to 70% of narcoleptic patients. The challenge in recognizing cataplexy is that normal subjects can endorse cataplexy-like symptoms. The prudent clinician should then inquire about the presence of cataplexy with a vague, initial question such as, “Does anything unusual happen when you tell a joke or a funny story, hear something funny or laugh, or make some witty verbal remark?” It is often helpful for the clinician to ask patients to describe their first and last episode, inquiring about emotional triggers, muscle groups affected, duration, and frequency of attacks (Fig. 11.1-2). Awareness is maintained throughout the episode. Although difficult to ascertain, reflexes are abolished during a cataplectic episode. Recovery is usually complete and immediate, although abrupt transitions from wakefulness to sleep may follow an episode. Figure 11.1-2 Characterization of cataplexy: emotional triggers, muscle groups affected, duration, and frequency. Cataplexy typically starts in teens and adolescents. It usually manifests within 5 years of the onset of excessive sleepiness, although it can be delayed for more than 20 years after the onset of sleepiness. Interestingly, cataplexy symptoms often abate with advancing age, which may reflect the ability of the individual to learn and subsequently avoid triggers for cataplexy, or it may show the effectiveness of treatment. In children, however, cataplexy can occur without any recognizable trigger; these events are referred to as “cataplectic facies” (Fig. 11.1-3), which may result in facial muscle weakness, dropped eyelids and facial grimaces with the eyes kept open, facial slackening, mouth opening and/or tongue protrusion, and a sort of “drunken, droopy look” (Video 11-1). In a rare manifestation of cataplexy, so-called status cataplecticus, the episodes may persist for hours and may confine the patient to bed. Rebound cataplexy, which may result in status cataplecticus, occurs after the abrupt discontinuation of an antidepressant and usually results in more intense and frequent cataplectic attacks. Figure 11.1-3 Cataplexy. The pathophysiologic mechanism of KLS remains unknown, but imaging studies have revealed widespread abnormalities that include the hypothalamus and thalamus, possibly mediating sleepiness, and other cortical areas (Fig. 11.1-4). Treatment is largely supportive, but stimulants and mood stabilizers may be beneficial in some cases. Figure 11.1-4 Coregistered positron emission tomography (PET)/magnetic resonance images (MRI) from a patient with Kleine-Levin syndrome during a symptomatic and asymptomatic period. Our understanding of the pathophysiology of CNS hypersomnias is mostly limited to narcolepsy. The cause of narcolepsy was discovered as a result of research in canines and rodents (see Fig. 11.1-3). In humans, narcolepsy with cataplexy is due to the loss of hypocretin neurons in the posterior hypothalamus (Fig. 11.1-5). Hypocretin-1 is deficient in the cerebrospinal fluid (CSF) in most patients who have narcolepsy with cataplexy and in some cases of secondary narcolepsy and neurologic disorders. In contrast, CSF hypocretin levels are normal with IH and periodic hypersomnias. Figure 11.1-5 Hypocretin peptides and neurons in the lateral hypothalamus of narcoleptics versus controls and cerebrospinal fluid hypocretin levels in central nervous system hypersomnias and other conditions. In most cases of narcolepsy, mounting environmental and genetic evidence suggests that an autoimmune mechanism underlies the destruction of the hypocretin cell neurons (Fig. 11.1-6, A to D). As for many other autoimmune diseases, infections have been identified as potential triggers for narcolepsy, such as group A Streptococcus and influenza A. Seasonality in the incidence of narcolepsy has been well documented. Titers for antistreptolysin (ASO) antibodies, a marker for recent Streptococcus infection, are elevated in early-onset narcolepsy cases compared with late-onset cases. Most interestingly, the 2009 pandemic H1N1 outbreak was associated with an increased incidence of narcolepsy in children, and some cases developed after H1N1 vaccination with Pandremix. Figure 11.1-6 Environmental triggers and genetic factors in narcolepsy. In addition to environmental triggers for the development of narcolepsy, clear genetic risk factors for its development also exist. Monozygotic twins are 25% to 32% concordant for narcolepsy, and first-degree relatives of people with narcolepsy carry a 1% risk of developing the disorder. A major susceptibility factor for narcolepsy is a specific allele for one of the many human leukocyte antigen (HLA) genes located on human chromosome 6. HLA genes encode for specific protein complexes (major histocompatibility complexes) found on the surface of immune cells that present antigens to other immune cells, such as T cells (antigen-presenting cells [APCs]). DQB1*06:02, an allele of the HLA gene DQB1, is the most specific genetic marker for narcolepsy and is found in almost 99% of cases with hypocretin deficiency, which are typically associated with cataplexy, versus 12% to 38% of the general population, depending on ethnicity (Fig. 11.1-7). In addition to HLA, polymorphisms in the T-cell receptor (TCR) α-locus, P2RY11 purinergic receptors, OX40, and Cathepsin-H, which are all involved in immune processes, increase the susceptibility for narcolepsy (see Fig. 11.1-6, E). Figure 11.1-7 Human leukocyte antigens (HLA) involved in narcolepsy susceptibility. These findings suggest that a single peptide unique to hypocretin cells is mistakenly recognized by the immune system as foreign via several proposed immune pathways, leading to the autoimmune destruction of the hypocretin cell neurons (Fig. 11.1-8). In contrast, secondary cases of narcolepsy, referred to as narcolepsy due to a medical condition in the ICSD-2, may be due to a direct insult to the hypocretin system, rather than an autoimmune mechanism, a theory supported by the fact that most tumors associated with narcolepsy are located in the hypothalamus (Fig. 11.1-9). In other cases, narcolepsy may be the result of complex pathology, whether associated with cataplexy or hypocretin deficiency or neither, including myotonic dystrophy; ataxia, deafness, narcolepsy, and dementia; Prader-Willi syndrome; Niemann-Pick disease; and various other pathologies. Figure 11.1-8 Potential pathways for a role of influenza A or streptococcal infections in the development of the autoimmune destruction of hypocretin cells. Figure 11.1-9 Neurologic diseases and location of brain lesions in 113 cases of secondary narcolepsy. Although hypocretin deficiency has been identified as the cause of narcolepsy, the pathophysiologic mechanisms underlying sleepiness, cataplexy, and other REM-related phenomenon in narcolepsy are complicated and, at times, controversial. A simplistic model depicts the hypocretin system sending widespread projections throughout the CNS, such as monoaminergic nuclei to promote wakefulness and motor neurons and the ventrolateral periaqueductal gray matter/lateral pontine tegmentum (vlPAG/LPT) to maintain muscle tone (Fig. 11.1-10). Figure 11.1-10 A theoretic and simplified model for the hypocretin system in the promotion of wakefulness and control of muscle tone. The clinician must first differentiate sleepiness from fatigue and exhaustion. The severity of the symptom can then be further characterized with subjective scales such as the Epworth Sleepiness Scale (see Chapter 8, Fig. 8-16) and the Fatigue Severity Scale (Box 11.1-2). A PSG is routinely performed to characterize sleep architecture and evaluate for sleep disorders such as SDB and periodic limb movements during sleep (PLMS). In narcolepsy/hypocretin deficiency, the PSG typically shows sleep fragmentation, increased stage 1 sleep, and, in 50% of cases, a sleep-onset REM period (SOREMP), defined as a REM latency of 15 minutes or less (Fig. 11.1-11). A SOREMP on the PSG is diagnostic for narcolepsy, but the PSG shows no findings diagnostic for IH. A high sleep efficiency and/or prolonged total sleep time on the PSG in the proper clinical context may be suggestive of IH. Figure 11.1-11 A sleep-onset rapid eye movement period during an overnight polysomnogram performed in a patient with excessive daytime sleepiness and cataplexy. If the overnight PSG is not diagnostic for a sleep disorder to account for the EDS, then a multiple sleep latency test (MSLT), which measures the tendency of a patient to fall asleep, or, alternatively, a maintenance wakefulness test (MWT), which measures the ability to maintain wakefulness, is performed. The MSLT is a series of four to five naps across the day taken in 2 hour intervals and is typically used for diagnostic purposes, whereas the MWT is a series of four 40-minute trials across the day in 2-hour intervals, routinely performed to document the ability of a person (e.g., an airline pilot) to maintain wakefulness after a prescribed therapy, such as after the successful treatment of SDB with a continuous positive airway pressure (CPAP) machine. During each nap of an MSLT, the patient is instructed to try to fall asleep and is given 20 minutes to do so; if no sleep is observed, the “nap” is ended. If the patient falls asleep, the nap continues for another 15 minutes, and the presence or absence of REM sleep (SOREMP) in each nap is recorded (Fig. 11.1-12). A regular sleep schedule and adequate sleep time, ideally documented with a sleep diary and/or actigraphy, should be maintained for 2 weeks before the MSLT. Medications that affect the ability to fall asleep, such as stimulants, or those that affect the propensity to enter REM sleep, such as antidepressants, should be discontinued for at least 2 weeks before the MSLT. Ideally, a blood or urine drug screen should be performed on the day of the MSLT. Most of the technical procedures of the MWT and MSLT are similar, except in the MWT, the subject is instructed to try to stay awake. The MWT trial is terminated after maintaining wakefulness for 40 minutes or after the first epoch of unequivocal sleep. Figure 11.1-12 A multiple sleep latency test (MSLT) diagnostic for narcolepsy from the patient shown in Figure 11.1-11. Treatment of CNS hypersomnias generally requires both behavioral and pharmacologic treatments. Behavioral treatments include the maintenance of a regular sleep schedule and naps. Educating the patient and family about the disease helps foster a supportive network (support groups, school adaptation) and treatment compliance. Pharmacologic management includes medications to treat EDS, cataplexy, and other REM-related phenomena and disrupted nocturnal sleep. Medications include stimulants, antidepressants, and sodium oxybate (Table 11.1-1). A conservative pharmacologic regimen is generally encouraged, particularly if the clinical picture or etiology is unclear or multifactorial in nature (e.g., narcolepsy without cataplexy or hypocretin deficiency) because many of these pharmacologic agents possess significant side effect profiles and come with a potential for abuse. In contrast, when cataplexy is present or hypocretin deficiency is documented, the disease is lifelong, and treatment is more codified and generally mandates aggressive optimization of medications. Anic-Labat, S, Guilleminault, C, Kraemer, HC, et al. Validation of a cataplexy questionnaire in 983 sleep-disordered patients. Sleep. 1999; 22(1):77–87. Arnulf, I, Rico, TJ, Mignot, E. Diagnosis, disease course, and management of patients with Kleine-Levin syndrome. Lancet Neurol. 2012; 11:918–928. Dauvilliers, Y, Arnulf, I, Mignot, E. Narcolepsy with cataplexy. Lancet. 2007; 369:499–511. 2005. International Classification of Sleep Disorders: Diagnostic and Coding Manual. ed 2, Westchester, IL: American Academy of Sleep Medicine; 2005. Mignot, E. A practical guide to the therapy of narcolepsy and hypersomnia syndromes. Neurotherapeutics. 2012; 9:739–752. Mignot, E, Lammers, GJ, Ripley, B, et al. The role of cerebrospinal fluid hypocretin measurement in the diagnosis of narcolepsy and other hypersomnias. Arch Neurol. 2002; 59(10):1553–1562. Ohayon, MM, Dauvilliers, Y, Reynolds, CF, 3rd. Operational definitions and algorithms for excessive sleepiness in the general population: implications for DSM-5 nosology. Arch Gen Psychiatry. 2012; 69(1):71–79. 11.2 Movement Disorders in Sleep The second version of the International Classification of Sleep Disorders (ICSD-2) lists several sleep-related movement disorders (see Box 11.2-1). A summary of all movement disorders is shown in Figure 11.2-1. The ones most commonly experienced in sleep medicine are restless legs syndrome (RLS), also known as Willis-Ekbom disease, and periodic limb movement disorder (PLMD), which is the focus of this chapter. A brief discussion is also provided to cover three other sleep-related movement disorders that sleep clinicians may also encounter: sleep-related leg cramps, bruxism, and rhythmic movement disorder. Although hypnic jerks (Fig. 11.2-2), or “sleep starts,” are common and can have an extreme and almost violent phenomenologic appearance, they are physiologically normal and do not constitute a disorder. The remaining conditions listed as ICSD-2 sleep-related movement disorders are of limited, if any, clinical significance. Figure 11.2-1 Flowchart for the approach to the differential diagnosis of sleep-related movement disorders. The initial descriptions of periodic limb movements (PLMs) in sleep mistakenly classified these as abrupt, repetitive movements that represented a “nocturnal myoclonus,” a term used in the literature mostly before 1970, erroneously suggesting some relation to epileptic phenomena. These movements have now been appreciated as leg movements that can reflect variable characteristics—even within a single night, for one person—for duration, onset and offset patterns, periodicity, and even in muscle groups involved in the movements. The World Association of Sleep Medicine (WASM) developed the current generally accepted criteria for defining these movements (Fig. 11.2-3). As shown in Figure 11.2-4, these movements can be frequent; in some patients, they are associated with arousals from sleep. Because these limb movements can occur during wakefulness or sleep, they are referred to as PLMS when they occur during sleep and PLMW when they occur during wakefulness. Movements may also occur in the arms but generally are limited to the legs. Figure 11.2-3 Definition of periodic limb movements (PLMs). Sleep, and to some extent the drowsy resting state, inhibits spinal leg-flexor reflexes. This has been demonstrated for sleep-related inhibition of the leg-flexor response to high-frequency stimulation of the medial plantar nerve. PLMS may be associated with loss of this inhibition that occurs with aging and with some disorders, such as restless legs syndrome (RLS). Decreased spinal inhibition or, alternately phrased, increased spinal excitability, has been documented during sleep in patients with PLMS and RLS. The Hoffmann’s reflex inhibition that occurs for up to 0.5 seconds after it has been stimulated is reduced in patients with RLS during the evening awake resting time. This indicates increased spinal excitability for RLS that is limited to rest and to the time of day associated with RLS symptoms. In this commonly accepted view of PLMS, leg movements occur during sleep because of the failure to inhibit periodic descending spinal activation signals. Periodic descending activation events have been associated with the basic cycling alternating pattern (CAP) of sleep. CAP describes cycling between deeper (quiet) and more aroused (active) sleep, with PLMS occurring during the active cycle. PLMS and the associated activating events also occur with increases in blood pressure and heart rate that indicate autonomic arousal (Fig 11.2-5; see also Fig. 11.2-4). The periodic activating events, and not the PLMS, appear to be primary. Inhibiting the leg movements by dopaminergic drugs fails to alter CAP or these episodic autonomic activating events. Thus PLMS serves as a motor sign of failure to inhibit central nervous system (CNS)–activating events that have clinical and physiologic consequences, but the leg movements themselves do not cause the arousal events. Figure 11.2-5 Periodic limb movements (PLMs): clinical significance. PLMS and PLMW are age-related phenomena. PLMS occurs commonly in healthy adults older than 40 years, whereas PLMW occurs mainly in people younger than 19 years (Fig. 11.2-6) and to some extent in adults older than 60 years (Fig. 11.2-7). These differences in age effects suggest that neurobiologic origins differ for PLMW and PLMS. Figure 11.2-6 Polysomnogram in a 6-year-old child. The common occurrence of PLMS and PLMW in healthy individuals indicates that they are not sensitive markers for indicating any specific underlying disease state. They may, however, be a specific motor sign for some disease states, such as RLS, in which 80% to 90% of the patients have PLMS at rates greater than 5 per hour. PLMS and PLMW may act as indicators, albeit less sensitive, for both narcolepsy and rapid eye movement (REM) sleep behavior disorder. In addition, they can also result from medications such as selective serotonin reuptake inhibitors (SSRIs) or serotonin-norepinephrine reuptake inhibitors (SNRIs; Fig. 11.2-8). Figure 11.2-8 Certain antidepressants can worsen restless legs syndrome. PLMS that occurs at high rates for age with a sleep complaint unexplained by another disorder or medication has been assumed to define a sleep-related disorder referred to as periodic limb movement disorder (PLMD). Clinical guidelines for PLMD are given in Box 11.2-2. Despite considerable clinical experience that supports this condition, no studies have been done to clearly document this disorder. PLMS should be viewed first as markers of possible disease risk and is not a disorder itself. Some patients with high rates of PLMS and sleep complaints when treated with levodopa or a dopamine agonist developed RLS for the first time, indicating that PLMS may also occur as a forme fruste of RLS. RLS has been well defined by the International Restless Legs Syndrome Study Group (IRLSSG). The diagnostic criteria are available online at www.irlssg.org and are also listed in Box 11.2-3. The diagnosis requires all four of the basic essential criteria that define the disorder (Fig. 11.2-9) plus a differential diagnosis that excludes common RLS “mimics.” These mimics produce symptoms close to those of RLS that can be differentiated from RLS by careful history taking. Indeed, patients may sometimes find it difficult to articulate symptoms of RLS (Fig. 11.2-10), which may be one reason why the condition may not be very easily recognized by practitioners who first encounter patients with this condition. Several conditions need to be considered in the differential diagnosis of RLS (Table 11.2-1). The core symptom of RLS is the sensation of a strong urge to move the legs that occurs while resting, particularly in the evening or night. Thus RLS has a sensory disturbance with a movement response that sometimes occurs involuntarily. It is therefore considered a sensorimotor neurologic disorder precipitated by rest or sleep. Box 11.2-4 summarizes key clinical features for RLS diagnosis. Box 11.2-3 Diagnostic Criteria for Restless Legs Syndrome International Restless Legs Syndrome Study Group Essential Diagnostic Criteria (All Must Be Met) 1. An urge to move the legs usually, but not always, accompanied by or felt to be caused by uncomfortable and unpleasant sensations in the legs.*† 2. The urge to move the legs, and any accompanying unpleasant sensation, begins or worsens during periods of rest or inactivity, such as lying down or sitting. 3. The urge to move the legs, and any accompanying unpleasant sensation, is partially or totally relieved by movement, such as walking or stretching, at least as long as the activity continues.‡ 4. The urge to move the legs, and any accompanying unpleasant sensation, during rest or inactivity only occurs or becomes worse in the evening or night rather than during the day.§ 5. Occurrences of the above features are not solely accounted for as symptoms primary to another medical or a behavioral condition (e.g., myalgia, venous stasis, leg edema, arthritis, leg cramps, positional discomfort, habitual foot tapping.) Specifier for Clinical Significance of Restless Legs Syndrome The symptoms of RLS cause significant distress or impairment in social, occupational, educational, and other important areas of functioning by the impact on sleep, energy/vitality, daily activities, behavior, cognition, and mood. *Sometimes the urge to move the legs is present without the uncomfortable sensation, and sometimes the arms or other parts of the body are involved in addition to the legs. †Pediatric cases must include the uncomfortable and unpleasant leg sensation. ‡When symptoms are very severe, relief by activity may not be noticeable but must have been previously present. §When symptoms are very severe, the worsening in the evening or night may not be noticeable but must have been previously present. ¶The clinical course criteria do not apply for pediatric cases, nor do they apply for some special cases of provoked RLS, such as pregnancy- or drug-induced RLS, in which the frequency may be high, but it is limited to the duration of the provocative condition. Table 11.2-1 Differential Diagnosis of Restless Legs Syndrome (RLS) SNRIs, serotonin-norepinephrine reuptake inhibitors; SSRIs, selective serotonin reuptake inhibitors. Figure 11.2-9 Essential criteria and core symptoms of restless legs syndrome: “URGE.” (Modified from Allen RP, Picchietti D, Hening WA, et al: Restless legs syndrome: diagnostic criteria, special considerations, and epidemiology. A report from the Restless Legs Syndrome Diagnosis and Epidemiology Workshop at the National Institutes of Health. Sleep Med 2003;4[2]:101-119; and Walters AS: Toward a better definition of the restless legs syndrome. Move Disord 1995;10:634-642.) Figure 11.2-10 Patients sometimes find it difficult to articulate symptoms of restless legs syndrome. Serum ferritin and transferrin saturation should be obtained in all RLS patients; those with ferritin level below 75 µg/L or transferrin saturation below 17% should be placed on oral iron treatment, provided that iron supplementation is not contraindicated, such as with hemochromatosis. Iron treatment should be stopped when ferritin exceeds 100 µg/L or transferrin saturation is 50% or greater. Patients with iron deficiency anemia as well as other comorbidities (Fig. 11.2-11) are at risk for developing RLS. Figure 11.2-11 Restless legs syndrome (RLS) in iron deficiency. RLS biology provides important insights into treatment options that involve three major contributors to the disease: iron, dopamine, and glutamate. In addition, genome-wide association studies have now identified allelic variations that carry increased risk of RLS (Table 11.2-2). One of these variations, BTBD9, has been found to be related to the presence of reduced serum ferritin, but otherwise, at this point these allelic variations have not been found to relate to any particular biologic pathway that increases the risk of RLS. Table 11.2-2 Major Allelic Variations Identified by Genome-Wide Association Studies as Increasing the Risk of Restless Legs Syndrome From Winkelmann J, Czamara D, Schormair B, et al: Genome-wide association study identifies novel restless legs syndrome susceptibility loci on 2p14 and 16q12.1. PloS Genet 2011;7(7):e1002171. Decreased iron content in the substantia nigra in patients with RLS has been documented by magnetic resonance imaging (MRI), ultrasound, and autopsy (Fig. 11.2-12). It is the best-documented biologic abnormality of RLS (Fig. 11.2-13). Iron in the brain is regionally regulated, and certain areas appear sensitive to decreased availability of iron, particularly the substantia nigra. RLS patients appear to have trouble maintaining adequate brain iron stores, possibly related to iron transport disturbance across the blood-brain barrier or failure of cellular distribution within the brain. This makes the brain iron stores particularly vulnerable to fluctuations in the peripheral iron state, and these individuals are more affected by low peripheral iron stores. Figure 11.2-14, A, shows the decreased relation between cerebral spinal fluid and serum ferritin for RLS patients compared with healthy adults without RLS. Accordingly, a critical feature of all RLS treatment is to maintain high normal levels of peripheral iron stores (i.e., serum ferritin >75 µg/L) to help augment the central iron stores and thereby reduce RLS severity (see Fig. 11.2-14, B). Future treatment development will focus on better methods of iron delivery for RLS, such as intravenous iron with formulations that promote iron transport to brain tissue. Figure 11.2-12 Restless legs syndrome (RLS) pathophysiology.
Neurologic Disorders
Classification and Prevalence
Clinical Features and Epidemiology
Narcolepsy can be described as a “pentad of symptoms” that includes sleepiness, cataplexy, sleep paralysis, hallucinations, and disrupted nocturnal sleep; it is uncommon for a patient to manifest all symptoms. RBD, rapid eye movement (REM) sleep behavior disorder; PLMS, periodic limb movements in sleep; OSA, obstructive sleep apnea.
Cataplexy
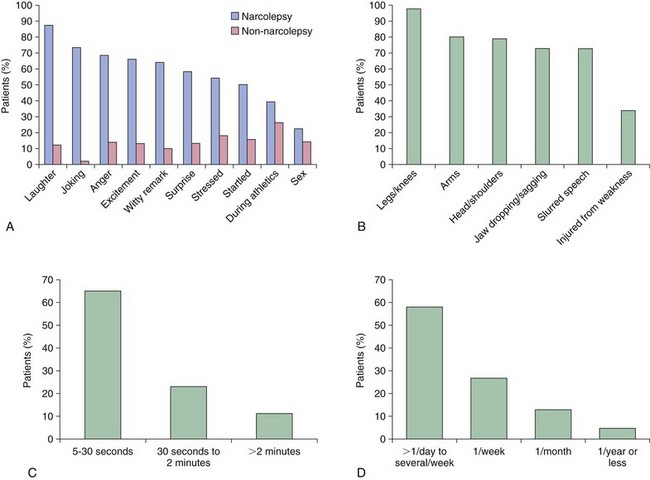
A, Comparison of emotional triggers for typical cataplexy and cataplexy-like episodes (e.g., physiologic muscle weakness) in narcoleptics (n = 63) and nonnarcoleptic subjects (n = 416/909). Surprisingly, cataplexy-like symptoms were claimed by 46% of the nonnarcolepsy subjects. Cataplexy is best differentiated from other types of muscle weakness when triggered by only three typical situations: when hearing or telling a joke, while laughing, or when angry. B, Muscle groups affected in typical cataplexy. The most commonly affected muscle groups involve the legs/knees. A typical episode is often indiscernible from normal behavior by an observer; it results in the knees buckling, arms dropping to the sides, slurred speech, and/or a slight dropping of the jaw. C, Duration of cataplexy episodes. Cataplexy typically lasts from a few seconds to nearly 30 seconds. D, Frequency of cataplexy episodes in narcolepsy cases. Episodes typically occur from once per day to several times per week. All subjects (n = 351) were human leukocyte antigen (HLA) DQB1*06:02 positive. (A, Modified from Anic-Labat S, Guilleminault C, Kraemer HC, et al: Validation of a cataplexy questionnaire in 983 sleep-disordered patients. Sleep 1999;22[1]:77-87. B, C, and D, Modified from Okun ML, Lin L, Pelin Z, Hong S, Mignot E: Clinical aspects of narcolepsy-cataplexy across ethnic groups. Sleep 2002;25[1]:27-35.)
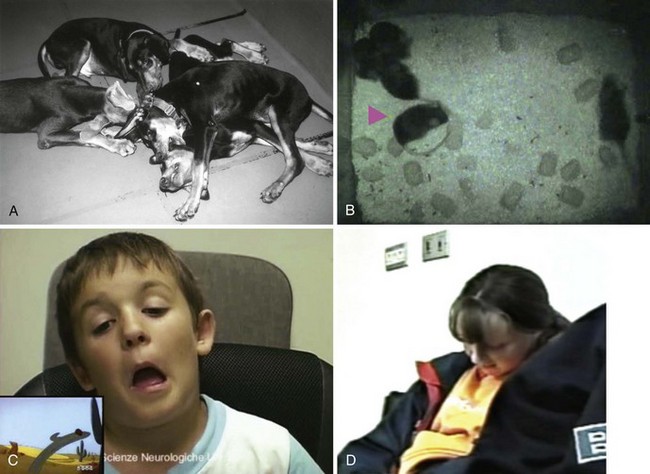

A, Cataplexy in Doberman Pinschers. B, Cataplexy in mice. C, “Cataplectic facies” in a child watching a cartoon. D, Head-dropping and slumping of a child in a chair. E, A cataplectic episode in an adult demonstrating buckling of the knees and falling to the floor. (A, Courtesy Emmanuel Mignot. B, From Scammell TE, Willie JT, Guilleminault C, Siegel JM: International Working Group on Rodent Models of Narcolepsy: A consensus definition of cataplexy in mouse models of narcolepsy. Sleep 2009;32[1]:111-116. C, From Serra L, Montagna P, Mignot E, Lugaresi E, Plazzi G: Cataplexy features in childhood narcolepsy. Mov Disord 2008;23[6]:858-865. D, From Macleod S, Ferrie C, Zuberi SM: Symptoms of narcolepsy in children misinterpreted as epilepsy. Epileptic Disord 2005;7[1]:13-17. E, From Overeem S, Mignot E, van Dijk JB, Lammers GJ: Narcolepsy: clinical features, new pathophysiologic insights, and future perspectives. J Clin Neurophysiol 2001;18[2]:78-105.)
Kleine-Levin Syndrome
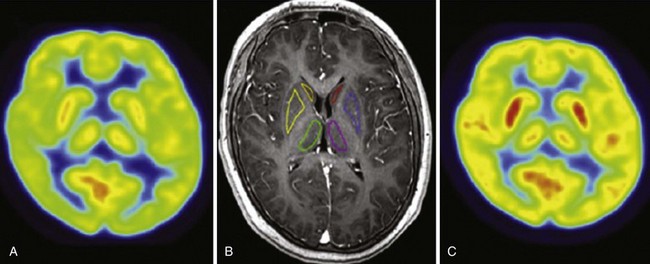
A, In a symptomatic phase, 18F-fluorodeoxyglucose (FDG)-PET demonstrates a decrease in glucose metabolism compared with the asymptomatic phase in the thalamus, hypothalamus, caudate nuclei, and striatum. These overall findings are similar to prior single-photon emission computed tomography findings. B, Regions of interest (bilateral caudate nuclei, striatum, and thalami) by MRI. C, FDG-PET in an asymptomatic phase. (Modified from Lo YC, Chou YH, Yu HY: PET findings in Kleine-Levin syndrome. Sleep Med 2012;13[6]:771-772.)
Pathophysiology
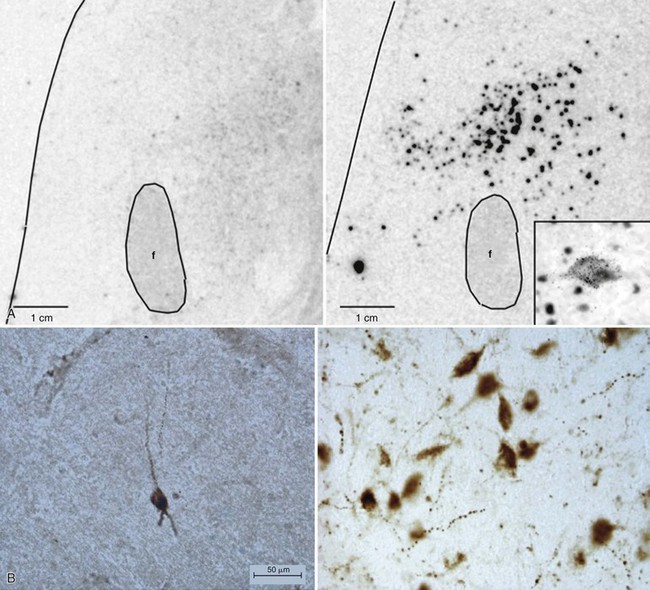
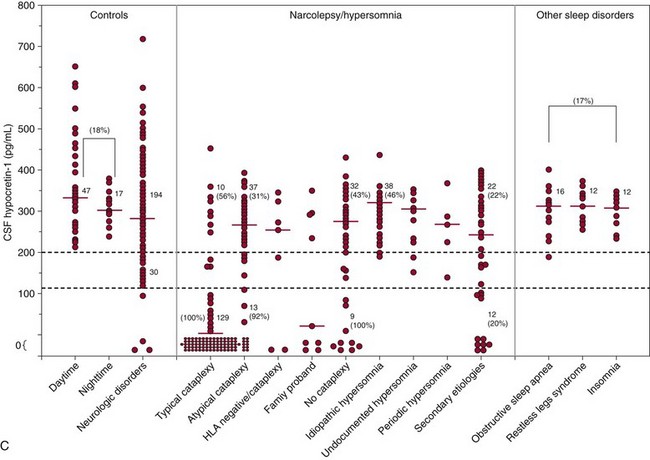
A, Dramatic reduction of hypocretin mRNA (gene expression) in the lateral hypothalamus in a narcoleptic (left) versus a control brain (right). B, Dramatic reduction of hypocretin-stained peptides (hypocretin cells) in the lateral hypothalamus in a narcoleptic (left) versus a control brain (right). Narcoleptics have an 85% to 95% reduction in the number of hypocretin neurons. f, fornix. C, Cerebrospinal fluid (CSF) hypocretin levels and HLA DQB1*06:02 status in controls, central nervous system hypersomnias, and other sleep disorders. Each point represents the crude concentration of hypocretin-1 (normal, >200 pg/mL; low, <110 pg/mL) in a single person, with the total number of subjects noted (digits without parentheses) in each range and median value, denoted with a horizontal bar in each group. HLA-DQB1*06:02 positivity is a percentage, denoted in parentheses. Normal hypocretin levels were noted in most of the controls and in all the periodic hypersomnias, idiopathic hypersomnias, and other sleep disorders. Almost all hypocretin-deficient narcolepsy cases are HLA DQB1*06:02 positive, whereas healthy control subjects are DQB1*06:02 positive in up to 38% of cases (18% in this figure). No consistent data are available for HLA DQB1*06:02 status in idiopathic hypersomnia and secondary narcolepsy. (A, Modified from Peyron C, Faraco J, Rogers W, et al: A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med 2000;6:991-997. B, Modified from Thannickal TC, Moore RY, Nienjuis R, et al: Reduced number of hypocretin neurons in human narcolepsy. Neuron 2000;27[3]:469-474. C, Modified from Nishino S, Mignot E: Narcolepsy and cataplexy. Handb Clin Neurol 2011;99:783-814.)
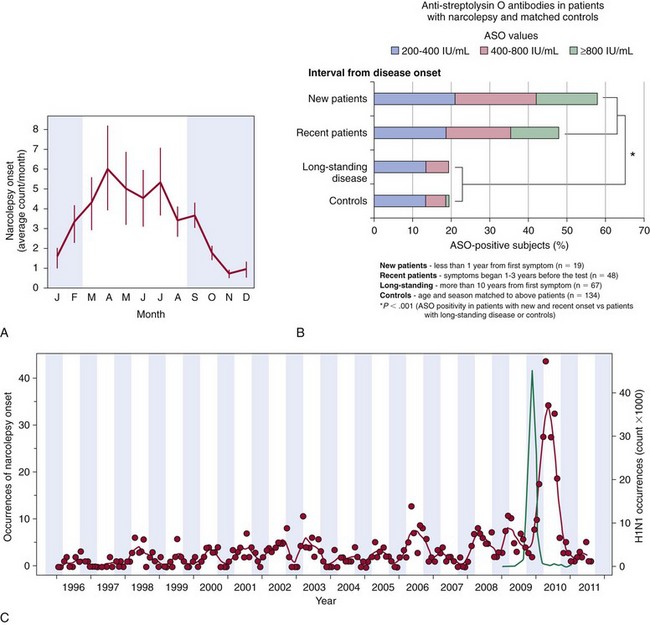

A, Seasonality and the incidence of narcolepsy in China. Data as mean ± standard error of the mean of monthly occurrences, corrected by the number of days per month and leap years, in percentage of 12 months across the year (mean of 15-year data). Onset of disease is approximately sixfold to sevenfold more frequent in the late spring and early summer versus late fall or early winter (shaded areas). B, Antistreptolysin O (ASO) antibodies in patients with narcolepsy with hypocretin deficiency (n = 200) and age-matched controls (n = 200). Higher ASO titers were seen in narcoleptics, particularly closer to disease onset, compared with controls. This suggests that streptococcal infections are probably a significant environmental trigger for narcolepsy. C, The 2009 H1N1 pandemic in China was associated with a threefold increase in narcolepsy onset. Monthly counts of onset occurrences over a 15-year period (raw data) with a 3-month moving average trend line are depicted in red (n = 629 diagnosed with narcolepsy/hypocretin deficiency at People’s Hospital, Beijing University, China). Note the clear seasonal fluctuations of the trend line with lower onset counts in fall and winter (shaded areas) and higher onset counts in spring and summer. Number of H1N1 infections documented by governmental statistics is depicted in green. The peak of infections was followed by a large increase in incident narcolepsy cases in 2010, which was largely independent of H1N1 vaccination (only 8 of 142 patients recalled receiving the vaccination), followed by an abrupt decline in both infections and narcolepsy cases in 2011. D, H1N1 vaccination with Pandremix was associated with an increased incidence of the diagnosis (Dx) of childhood narcolepsy in Sweden, Ireland, and Finland. All cases that underwent HLA typing were found to be HLA DQB1*06:02 positive. E, Known narcolepsy loci involved in immune processes and increased narcolepsy susceptibility. HLA DQB1*06:02, a specific allele for one of the many HLA genes located on human chromosome 6, is tightly associated with hypocretin deficiency. These HLA genes encode for major histocompatibility complexes that present antigens to other immune cells. Polymorphisms in the T-cell receptor (TCR)-α locus, P2RY11 purinergic receptors, TNFSF4 (formerly OX40), and CTSH (Cathepsin-H) are all involved in immunity, and all increase the risk of narcolepsy. TCR is the receptor for HLA antigen presentation on T cells. Purinergic receptors, which are known to have immune-modulatory effects—such as chemotaxis, maturation, and regulated cell death—are poorly understood. OX40, for example, is a co-stimulatory molecule for T- and B-cell activation. CTSH, a member of the papain-like family, participates in intracellular protein degradation. Mutations in DNMT1, a widely expressed DNA methyltransferase involved in development, have been identified in autosomal-dominant cerebellar ataxia, deafness, and narcolepsy (ADCA-DN). (A and C, Modified from Han F, Lin L, Warby SC, et al: Narcolepsy onset is seasonal and increased following the 2009 H1N1 pandemic in China. Ann Neurol 2011;70[3]:410-417. B, Modified from Aran A, Lin L, Nevsimalova S, et al: Elevated anti-streptococcal antibodies in patients with recent narcolepsy onset. Sleep 2009;32[8]:979-983. D, Data from Partinen M, et al: Increased incidence and clinical picture of childhood narcolepsy following the 2009 H1N1 pandemic vaccination campaign in Finland. PLoS One 2012;7(3):e33723. E, Courtesy Dr. Juliette Faraco.)
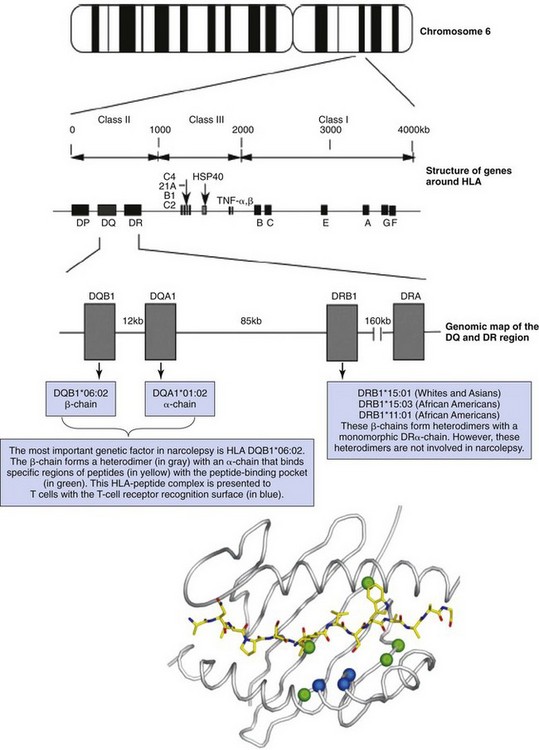
The HLA genes are located on chromosome 6 and are distributed over more than 4000 kilobases (kb). Antigen-presenting cells, macrophages, and dendritic cells have HLA class I (A, B, and C) and II (DQ, DR, and DP) protein; the locations of genes enclosing these proteins are shown. The HLA gene family is divided into two classes and is located in major histocompatibility complex (MHC) class I (A, B, and C) and class II (DQ, DR, and DP) regions. Close to the HLA genes in the MHC class III region are genes that encode complements 2 and 4, tumor necrosis factor (TNF), and heat shock protein (HSP40). The HLA DR and DQ genes are located very close to each other. HLA class II DR and DQ genes are heterodimers encoded by two genes each: one generates an α-chain, the other a β-chain. All these genes are located within a small genetic distance, leading to extremely high linkage disequilibrium. DRA is monomorphic and does not contribute significantly to HLA diversity, in contrast with DQA1, DQB1, and DRB1, which have several hundred possible alleles. The most important genetic factor in narcolepsy is HLA DQB1*06:02. In whites and Asians, the associated DR2 subtype DRB1*15:01 is typically observed with DQB1*06:02 (and DQA1*01:02) in narcoleptic patients. In African Americans, either DRB1*15:03, a DNA-based subtype of DR2, or DRB1*11:01, a DNA-based subtype of DR5, is observed most frequently, together with DQB1*06:02—the most specific marker for narcolepsy across all ethnic groups. DQB1*06:02 forms a heterodimer (in gray) with an α-chain that binds specific regions of peptides with the peptide-binding pocket (in green). This HLA-peptide complex is presented to T cells with the T-cell receptor recognition surface (in blue). (Modified from Nishino S, Mignot E: Narcolepsy and cataplexy. Handb Clin Neurol 2011;99:783-814; and Jones EY, Fugger L, Strominger JL, Siebold C: MHC class II proteins and disease: a structural perspective. Nat Rev Immunol 2006;6:271-282.)
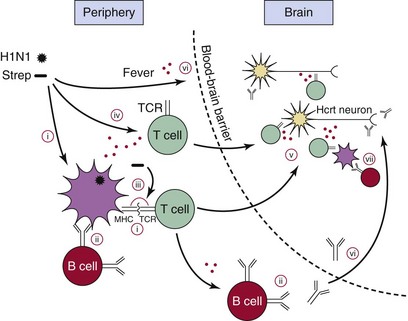
A peripheral H1N1 influenza or Streptococcus pyogenes (Strep) infection could stimulate autoreactive T cells or B cells via several different mechanisms. Selected resting autoreactive T cells and B cells may have reactivity toward hypocretin (Hcrt) cells, having escaped negative selection in the thymus. These could be activated in the following ways. (i) Molecular mimicry, T cells. Antigens from the virus or bacteria are presented, for example, by major histocompatibility complex (MHC) DQA1*01:02-DQB1*06:02 on an antigen-presenting cell (APC). The T cell recognizes the antigen and is activated. The same T cell, or a clone, migrates to the brain, where it recognizes an Hcrt cell–specific antigen (cross-reactivity), thereby inducing the autoimmune attack. (ii) Molecular mimicry, B cells. An autoreactive B cell can be activated if it also recognizes an antigen from the pathogen. This process requires signals from activated T cells (T-cell help). (iii) Superantigens from Streptococcus cross-link the MHC and T-cell receptor (TCR) molecules independent of antigen specificity, activating the autoreactive T cell. (iv) Bystander activation. Resting autoreactive cells are activated as a result of general immune activation independent of specific antigens. (v) Lymphocyte migration to the central nervous system (CNS). Once activated, the T cells can migrate to the brain. Depending on the type of T cell, a variety of mechanisms could account for the autoimmune attack. MHC class II expression is restricted to microglia, but MHC class I molecules are expressed in various brain cells, including neurons. (vi) Opening of the blood-brain barrier. Fever and other factors associated with the general immune response allow lymphocytes to penetrate the blood-brain barrier more easily and also allow antibodies to access the CNS. (vii) Production of autoantibodies. This can also occur as a secondary response to Hcrt cell death via APCs from the brain that have phagocytosed the dead neurons. Red dots indicate processes in which release of cytokines or cytotoxic substances plays an important role. H1N1, H1N1 influenza A virus or epitopes from adjuvanted vaccines. (Modified from Kornum BR, Faraco J, Mignot E: Narcolepsy with hypocretin/orexin deficiency, infections, and autoimmunity of the brain. Curr Opin Neurobiol 2011;21:897-903.)
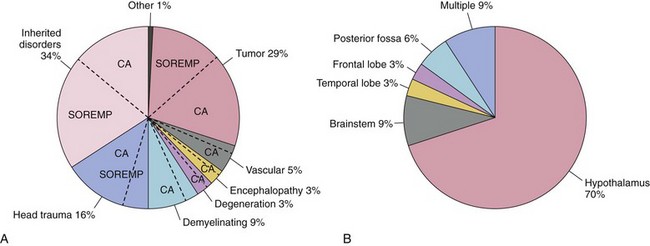
A, Neurologic diseases by category reported as secondary narcolepsy. Tumors, inherited disorders, and head trauma are the three most frequent causes. The percentage of cataplexy (CA) or sleep-onset rapid eye movement periods (SOREMP) is denoted in each category with a dashed line. B, Location of brain lesions in symptomatic cases with narcolepsy associated with brain tumor; lesions in the hypothalamus and adjacent structures are the most common location. Included are 113 symptomatic cases of narcolepsy. (Modified from Kanbayashi T, Sagawa T, Takemura F, et al: The pathophysiologic basis of secondary narcolepsy and hypersomnia. Curr Neurol Neurosci Rep 2011;11[2]:235-241.)
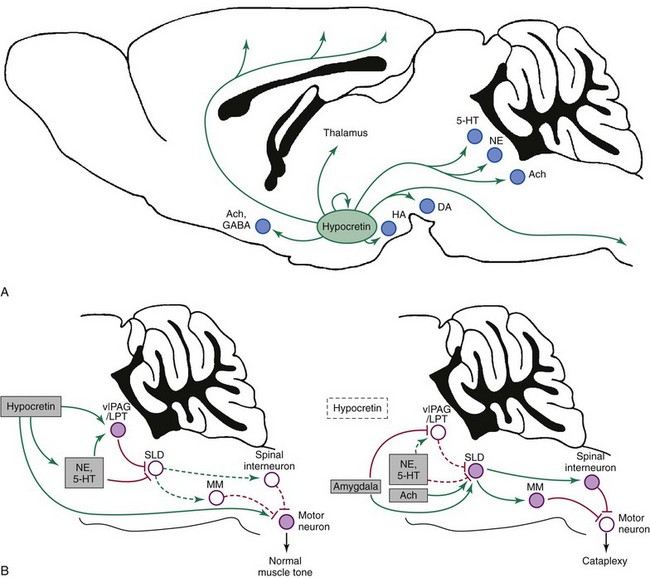
A, Hypocretin neurons in the lateral hypothalamus innervate wake-promoting nuclei, including neurons that produce histamine (HA, tuberomammillary nucleus), norepinephrine (NE, locus ceruleus), serotonin (5-HT, dorsal raphe), dopamine (DA, ventral tegmental area), and acetylcholine (Ach, basal forebrain, pedunculopontine, and laterodorsal tegmental nuclei). In addition, the hypocretin neurons provide direct, excitatory inputs to the cortex, thalamus, and spinal cord. GABA, γ-aminobutyric acid. B, Several pathways suppress atonia during normal wakefulness (left). Atonia is driven by neurons in the sublaterodorsal tegmental nucleus (SLD) that activate neurons in the spinal cord and medial medulla (MM) that inhibit motor neurons using GABA and glycine. During wakefulness, this atonia system is inhibited by neurons in the ventrolateral periaqueductal gray matter/lateral pontine tegmentum (vlPAG/LPT) and by monoaminergic neurons, such as NE and 5-HT. The hypocretin neurons are active during wakefulness and are silent during rapid eye movement sleep. They help maintain normal muscle tone by exciting monoamine neurons, motor neurons, and neurons in the vlPAG/LPT. In narcolepsy (right), the loss of the hypocretin neurons plus strong, positive emotions can trigger cataplexy. Positive emotions may activate neurons in the amygdala that excite the SLD and inhibit the vlPAG/LPT. The SLD may also be activated by cholinergic inputs and a sudden withdrawal of monoamine tone. The SLD then excites neurons in the medial medulla and spinal cord that strongly hyperpolarize motor neurons, resulting in cataplexy. Normally, the effects of the hypocretin system and a continued monoaminergic drive to the pons and directly to motor neurons would counter this triggering of atonia, but in the absence of hypocretin, these excitatory drives are lost, and cataplexy occurs. Solid pathways from filled nuclei are active; dashed pathways from unfilled nuclei are inactive. Green pathways are excitatory; red pathways are inhibitory. (Modified from Burgess CR, Scammel TE: Narcolepsy: neural mechanisms of sleepiness and cataplexy. J Neurosci 2012;32[36]:12305-12311.)
Evaluation and Diagnosis
Epworth Sleepiness Scale and Fatigue Severity Scale
Overnight Polysomnography, Multiple Sleep Latency Test, and Maintenance of Wakefulness Test
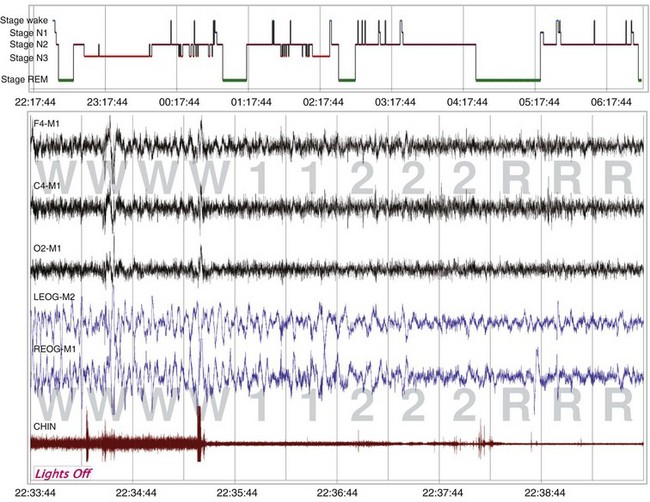
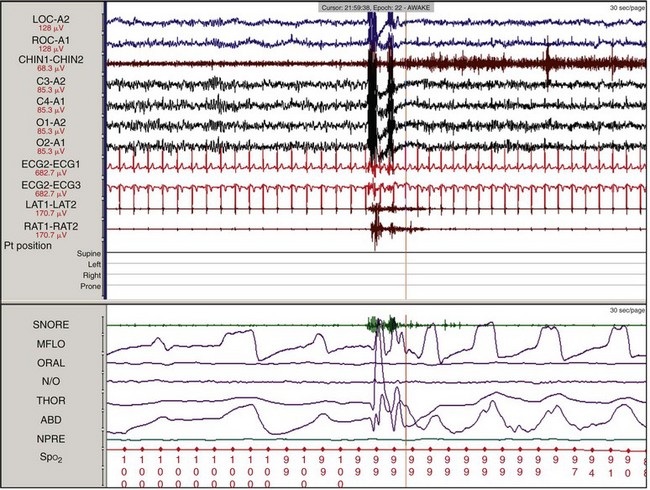
The sleep hypnogram (top) and selected fragments from epochs of the recording (bottom), starting from “lights out,” demonstrate the rapid onset of rapid eye movement (REM) sleep. A normal REM latency is typically defined as 90 to 120 minutes; in this recording, it is 2.5 minutes. A REM latency of 15 minutes or less is diagnostic for narcolepsy.
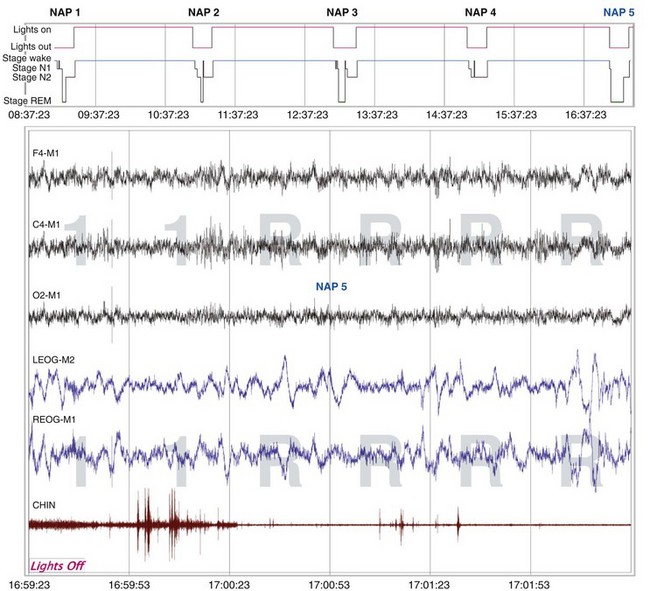
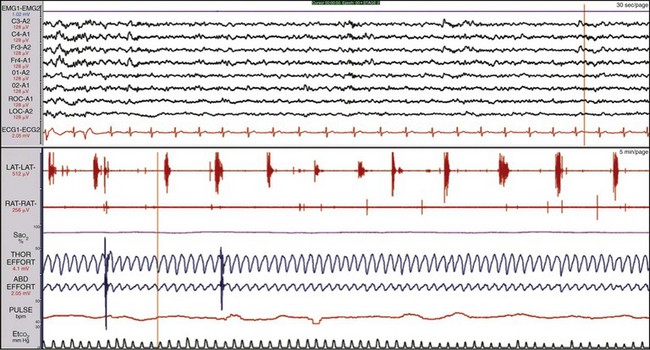
The sleep hypnogram (top) shows that the patient attained rapid eye movement (REM) sleep on four of the five naps. Selected epochs from the NAP 5 recording (bottom), starting from “lights out,” show the patient immediately falling asleep (sleep latency, 0 min) and entering REM sleep 1 minute later (REM latency, 1 min). The calculated mean sleep latency of all five naps was 1.6 minutes. Given the MSLT findings and sleep onset REM period (SOREMP) on the polysomnogram, the patient was diagnosed with narcolepsy with cataplexy.
Treatment
Overview
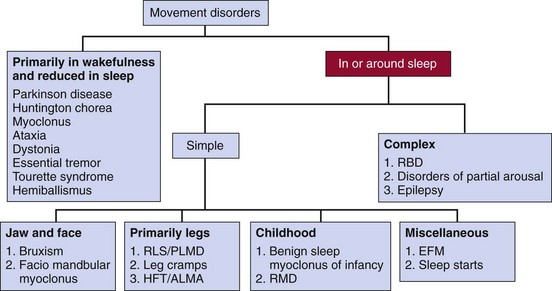
ALMA, alternating leg muscle activation; EFM, excessive fragmentary myoclonus; HFT, hypnagogic foot tremor; PLMD, periodic limb movement disorder; RBD, rapid eye movement (REM) sleep behavior disorder; RLS, restless legs syndrome; RMD, rhythmic movement disorder.
Periodic Limb Movements in Sleep and Periodic Limb Movement Disorder
History and Definition
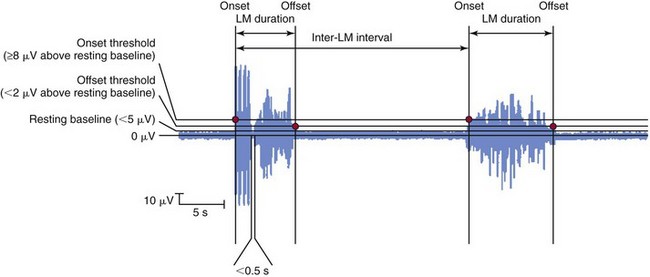
Movement starts when the electromyogram exceeds the onset threshold and ends when it remains below the offset threshold for at least 0.5 seconds. The leg movement duration must be 0.5 to 10 seconds, and the intermovement interval must be 5 to 90 seconds. Four consecutive movements (three intermovement intervals) must be observed for these to be counted as PLMs. The number of such movements divided by the observation time is expressed as the PLM index, usually in terms of PLMs per hour. LM, limb movement. (From Zucconi M, Ferri R, Allen R, et al: The official World Association of Sleep Medicine [WASM] standards for recording and scoring periodic leg movements in sleep [PLMS] and wakefulness [PLMW] developed in collaboration with a task force from the International Restless Legs Syndrome Study Group [IRLSSG]. Sleep Med 2006;7:175-183.)
Biology and Pathophysiology
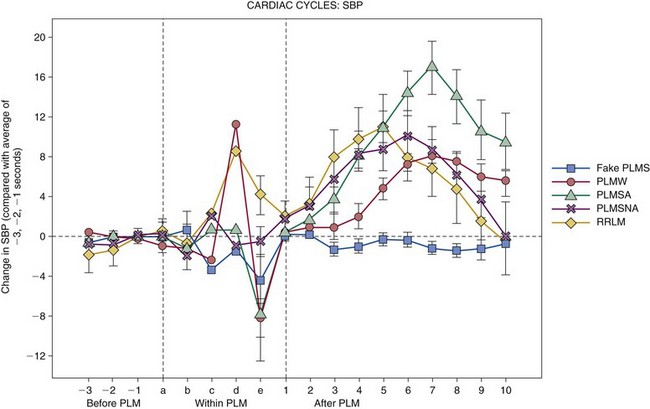
PLMs are common in healthy older adults but are not disease state specific. They are a motor sign of restless legs syndrome, with 80% to 90% sensitivity (not specific), and are associated with heart rate and blood pressure increases. PLMS, PLM during sleep; PLMW, PLM during wakefulness; PLMSA, PLMs with cortical arousal; PLMSNA, PLMs without cortical arousal; RRLM, respiratory-related limb movements; SBP, systolic blood pressure. (From Siddiqui F, Strus J, Ming X, et al: Rise of blood pressure with periodic limb movements in sleep and wakefulness. Clin Neurophysiol 2007;118:1923-1930.)
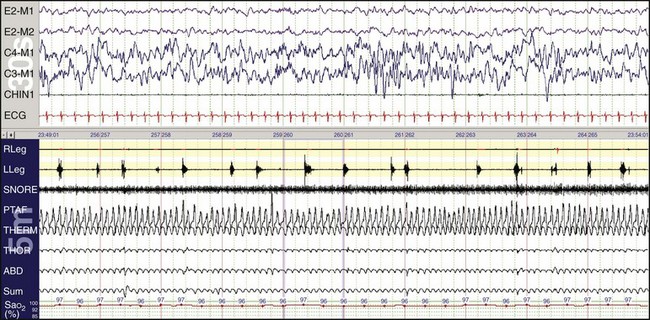
Children can have periodic limb movements during sleep. Such children may be referred with sleepiness and may have been diagnosed with attention deficit–hyperactivity disorder. In this example, inadequate gain resulted in the right leg not showing the movements.
Clinical Significance
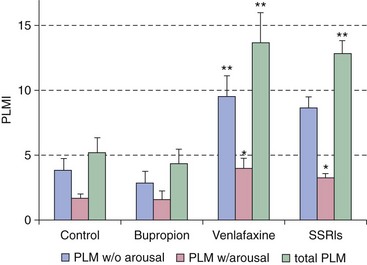
In this example, the serotonin-norepinephrine reuptake inhibitor venlafaxine (Effexor) is a risk factor for inducing periodic limb movements with and without arousals compared with control patients, those using selective serotonin reuptake inhibitors (SSRIs), and those taking bupropion (Wellbutrin). PLMI, periodic limb movement index. *P < .001; **P < .0001. (Modified from Yang C, White DP, Winkelmann JW: Antidepressants and periodic leg movements of sleep. Biol Psychiatry 2005;58:510-514.)
Clinical Guidelines
Restless Legs Syndrome
Diagnosis
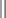
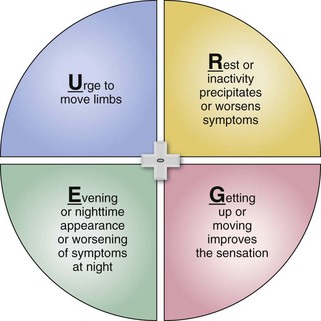
Patients speak in emotional language. (Has the term “heebie-jeebies” ever been noted in a diagnostic scale or evidence-based scientific paper?) Physicians look for top-line factual descriptors about restless legs syndrome and often do not interpret what the patient is saying.
Medical Evaluation: Iron Status
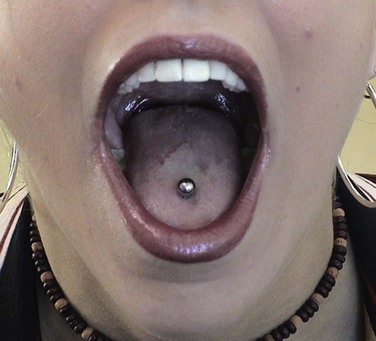
This young woman with RLS was a vegetarian on a poor diet and had severe, chronic iron deficiency. Her tongue had evidence of glossitis manifested as reddened atrophic mucosa. Burning of the tongue is a common symptom when this is present.
Biology and Pathophysiology

Iron.
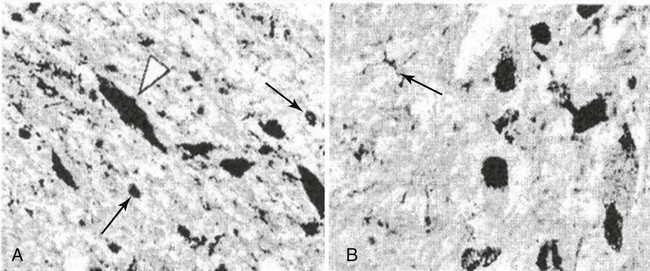
Brain iron insufficiency neuropathologic examination using iron stain for RLS controls (A) and patients (B). (From Connor JR, Boyer PJ, Menzies SL, et al: Neuropathological examination suggests impaired brain iron acquisition in restless legs syndrome. Neurology 2003;6:301-309.)![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree