Unlike most chapters in this book, which focus on conditions that affect the central nervous system (CNS), this one focuses on conditions that affect the peripheral nervous system (PNS). While the CNS consists of the brain and spinal cord, the PNS consists of the nerves, muscles, and neuromuscular junctions (NMJ). Neurologists refer to conditions that affect the PNS as neuromuscular disorders. Some disorders affect both the CNS and PNS and may lead to changes in cognition, behavior, or emotion in addition to signs and symptoms of PNS disease—as highlighted in this chapter.
Children and adolescents with neuromuscular disorders typically come to the attention of psychiatrists for one or more of three reasons. First, psychiatrists can play a crucial role in the management of emotional or behavioral manifestations or sequelae of the disorder. In particular, children with neuromuscular disorders can suffer from anxiety disorders, depression, or significant behavioral problems that may or may not be related to cognitive disability.
Second, a psychiatrist can help family members cope with the psychosocial consequences of disorders that potentially impact children’s behavior, coping mechanisms, and functional status. Hereditary diseases can be particularly disruptive to normal family dynamics. The parents of an affected child may have unresolved guilt related to passing on a genetic illness to their child and may also feel conflicted about having additional children. Siblings may have concerns about their own risk of developing the disease or may feel neglected because the affected child seems to receive a disproportionate amount of parental attention. Psychiatrists can help parents or siblings by providing psychotherapy and, when needed, pharmacotherapy.
Finally, psychiatrists acquainted with neuromuscular conditions may aid in proper diagnosis. For example, patients with neuromuscular disease are sometimes misdiagnosed as “psychogenic” and referred for psychiatric care. Such a misdiagnosis is especially likely when patients’ symptoms fluctuate or appear episodically, as in myasthenia gravis (MG) or periodic paralysis. A knowledgeable, astute psychiatrist may be able to identify dysfunction of the nervous system as an organic condition and ensure that the patient receives the appropriate treatment.
The majority of neuromuscular conditions exclusively affect the PNS and spare the CNS. Consequently, cognition is normal in most of these disorders, and the prevalence of major psychiatric illness is not significantly elevated. For example, children with polio, spinal muscular atrophy, and MG do not have cognitive impairment or psychiatric disturbance directly attributable to their condition—despite potentially devastating impairment in motor function. In contrast, weakness due to cerebral dysfunction (as in cerebral palsy) is often accompanied by cognitive abnormalities.
Anatomy and Physiology of the Peripheral Nervous System
The PNS transmits efferent information from the CNS to effectors, such as muscles and glands. It also carries afferent information to the CNS from the body and its environment. Disruption of efferent pathways often results in motor impairment, typically weakness. At the same time, damage to afferent pathways may produce sensory symptoms, particularly numbness and unpleasant sensations like spontaneously occurring pain.
The motor system controlling voluntary movement of skeletal muscle1 consists of two populations of neurons: the lower motor neurons (LMNs), which innervate muscles, and the upper motor neurons (UMNs), which terminate by forming synapses with the cell bodies of LMNs. The cell bodies of the UMNs are located in the motor strip of the cerebral cortex. Their axons form the corticospinal tracts, which descend through the white matter of the cerebral hemispheres and brainstem. In the medulla, the corticospinal tracts decussate (cross to the opposite side) and descend in the spinal cord.
On terminating in the spinal cord, each UMN axon synapses with the cell body of an LMN located in the anterior horn of the spinal gray matter. Axons of LMNs leave the spinal cord via the ventral nerve roots and then travel peripherally, joining fibers carrying afferent information to form a mixed spinal nerve. The motor axons continue peripherally in these nerves and ultimately end in a nerve terminal in close proximity to the skeletal muscle.
The nerve terminal contains synaptic vesicles, packets that hold the neurotransmitter acetylcholine. When an action potential propagated along a motor nerve reaches the nerve terminal, electrochemical changes (mediated by calcium ions) cause the vesicles to fuse with the presynaptic membrane and release acetylcholine into the NMJ. The acetylcholine diffuses across the synaptic cleft and binds to receptors in the postsynaptic membrane located on the muscle. This binding results in electrochemical changes (mediated by sodium and potassium ions) that trigger a muscle contraction.
In contrast to motor pathways, which enable us to move, the somatosensory system permits us to detect various physical stimuli that can be perceived as touch, pain, cold, vibration, and other sensations. Specialized organs that detect the stimuli, called receptors, are located in the skin and elsewhere. They convert the physical stimuli into electrical signals that travel toward the CNS along sensory fibers.2 These fibers join motor axons to form mixed nerves. The sensory axons carry afferent signals into the spinal cord via the dorsal roots. Sensory information travels to the brain chiefly through two tracts that ascend in the white matter of the spinal cord: the dorsal columns (which carry information about vibration, joint position, and light touch sensations) and the spinothalamic tracts (which mediate pain and temperature as well as light touch sensations).
In the lower cervical region, the mixed spinal nerves continue peripherally as the brachial plexus, a complex network of axons whose numerous branches provide motor and sensory innervation to the upper extremities (as well as frustration to students of anatomy). A similar structure, the lumbosacral plexus, innervates the lower extremities.
Principles of Diagnosis: Anatomic Localization
Disease processes can attack the PNS anywhere along its course. As they do when diagnosing CNS disease, neurologists try to localize a PNS lesion on the basis of the patient’s symptoms and signs. This section describes the relationships between various sites of pathology and the clinical patterns they produce.
Because lesions of either the CNS or PNS may produce sensory or motor signs and symptoms, neurologists usually begin their evaluation of a patient by trying to determine whether the pathology is in the CNS or PNS (or both). Pathology in the brain or spinal cord causes motor symptoms and signs characteristic of UMN dysfunction. Causes of such pathology are diverse; examples include stroke, trauma, tumor, and multiple sclerosis. In addition to weakness, UMN damage can also produce spasticity, increased deep tendon reflexes (DTRs), and Babinski signs. Moreover, lesions of the brain may also produce cognitive changes.
With lesions of the LMN, hypotonia (decreased muscle tone) and decreased or absent DTRs typically accompany weakness. The specific findings, of course, depend on the site of the lesion in the PNS (Table 18.1). Diseases that exclusively affect anterior horn cells, such as poliomyelitis or spinal muscular atrophy, result in atrophy, weakness, and loss of DTRs. Although these illnesses may cause devastating weakness, they do not produce cognitive impairment. Involuntary twitching movements of muscles, fasciculations, are most characteristic of these disorders of the anterior horn cell but can occur with any lesion of the LMN.
Injury of motor axons, which can result from a plethora of conditions, can occur anywhere along their course through nerve root, plexus, or peripheral nerve. Injuries may occur in single nerves or combinations of nerves. Moreover, because motor fibers travel with sensory fibers along much of their course, sensory abnormalities, such as pain, numbness, or tingling often accompany weakness.
TABLE 18.1 CLINICAL-ANATOMICAL LOCALIZATION OF NEUROMUSCULAR DISORDERS
Neurologists often differentiate abnormalities of peripheral nerve, neuropathies, from conditions affecting other segments of the axon. For example, nerve root involvement, or radiculopathy, may result from a herniated disc or an infection in the spinal subarachnoid space. Plexopathy involves the brachial or lumbosacral plexus, either completely or in a patchy distribution. Pathology in the root, plexus, or nerve typically causes muscle atrophy and weakness along with pain, numbness, or tingling. In addition, DTRs mediated by an affected nerve or nerve root will be depressed or absent.
Some processes such as focal entrapment affect an individual peripheral nerve (mononeuropathy). Generalized conditions such as diabetes can diffusely affect the PNS (polyneuropathy). Some conditions tend to affect several individual nerves (mononeuropathy multiplex).
Disruption of the NMJ, which occurs in botulism and MG, results in a pure motor syndrome without sensory features. It can cause generalized weakness or weakness restricted to one anatomical region. For example, MG often causes ptosis and weakness of extraocular muscles, but DTRs usually remain normal.
A wide variety of pathological processes may produce muscle disorders, myopathies. Neurologists recognize many causes of myopathy; etiologic categories include genetic, inflammatory, infectious, endocrine, metabolic, and toxic causes. The sections pertaining to specific diseases present details on each of these categories.
Approach to the Patient with Suspected Neuromuscular Disease
HISTORY
In neurology, as in psychiatry, the patient’s history remains the most important element of the evaluation. Although the examiner should endeavor to obtain as much information as possible directly from the patient, the child’s parents will often provide the majority of the information. The following components of the history deserve special mention.
Chief complaint
Children with neuromuscular disorders commonly present with weakness, numbness, tingling, gait difficulty, or ataxia. Neurologists seek to elicit a detailed description of the symptoms and should not accept at face value all terminology used by patients or their parents. For example, patients may state that they feel “weak” when in fact their problem is stiffness, rigidity, or incoordination. Similarly, because some terms, such as “numbness” and “dizziness,” have a variety of meanings, neurologists will ask a patient reporting such symptoms to describe them in greater detail. One technique is to ask patients and their parents to describe tasks that might be impeded by the symptoms.
Chronology of present illness
The time course of an illness often differentiates among etiologies. For example, trauma, infections, intoxications, and conversion reactions emerge acutely, over minutes to days. A subacute onset evolving over days to weeks typifies many systemic conditions, such as autoimmune, endocrine, and nutritional disorders. A chronic time course may suggest a genetic or metabolic abnormality. Because some conditions tend to progressively worsen while others are static, neurologists always try to determine if a patient’s symptoms are progressing or remaining stable. In addition, the patient’s age at the time the symptoms began should be ascertained, as many disorders have a typical age at onset.
As an example, consideration of the time course helps narrow the differential diagnosis of acute childhood paralysis. Hyperacute paralysis—developing over hours—is most often associated with hyperkalemia or hypokalemia or intoxication (such as with insecticides). It can also be psychogenic, perhaps triggered by an intolerable or disturbing thought or situation. In paralysis developing over days to weeks, Guillain-Barré syndrome (GBS) is the most common culprit. Other diagnoses to consider include transverse myelitis, botulism, MG, acute inflammatory diseases of muscle, infection of anterior horn cells by viruses (such as enteroviruses or, in endemic areas, polio), tick paralysis, and exposure to various neurotoxins. Rare causes include acute intermittent porphyria and diphtheria (1,2).
Medical history (including medications)
When approaching a patient with a possible neuromuscular disorder, neurologists attempt to identify comorbid medical conditions. Neuromuscular disorders may result from underlying medical illness, such as neuropathy due to leprosy, or from the treatment of medical illnesses, such as chemotherapy-induced neuropathy. Also, because some neuromuscular disorders involve other organs, evidence of such nonneurological involvement may serve as a clue to the diagnosis. Likewise, a single systemic illness may injure the PNS along with other systems. For example, MG may occur in conjunction with other autoimmune disorders.
Social history
Many substances of abuse, including alcohol, cocaine, and heroin, can damage the PNS. Glue sniffing, which is prevalent among adolescents, can cause neuropathy because users inhale toxic hexacarbons. Industrial exposures, to heavy metals for example, can also produce neuropathy.
Perinatal history
Physicians should note birth complications, including prematurity and large birth weight (macrosomia). Particularly when evaluating a hypotonic (floppy) infant, they should attempt to determine if fetal movements were decreased during the third trimester, suggesting that weakness was present in utero.
Development
Children with neuromuscular disorders may be delayed in attaining gross or fine motor milestones. On the other hand, coexisting impairment of language or other cognitive domains raises the likelihood of cerebral palsy or other brain pathology. Clinicians use instruments such as the Denver Developmental Screening Test (Denver Developmental Materials, Inc, Denver, CO) and its derivatives to assess whether children have achieved developmental milestones at the appropriate time and in the usual sequence.
Family history
Identifying an inherited disorder is critical for making an accurate diagnosis and providing genetic counseling. Physicians should ascertain whether family members are affected and, if so, attempt to determine the pattern of inheritance. Autosomal dominant conditions affecting the PNS include many types of hereditary neuropathy and certain myopathies, including myotonic dystrophy, facioscapulohumeral dystrophy, and one type of myotonia congenita. X-linked disorders include Duchenne and Becker muscular dystrophies and several varieties of hereditary neuropathy. Exclusively maternal transmission suggests a mitochondrial disorder, such as mitochondrial encephalomyopathy, lactic acidosis, and stroke (MELAS) or myoclonic epilepsy and ragged-red fibers (MERFF). Even if a child has no obviously affected family member, parental consanguinity may suggest that the child has a genetic disorder with autosomal recessive inheritance.
PHYSICAL AND NEUROLOGICAL EXAMINATION
Because some neuromuscular disorders are associated with systemic manifestations, physicians should perform a thorough general as well as neurological examination. They should always measure head circumference: an abnormally large head (macrocephaly) or a small head (microcephaly) suggests a cerebral cause for a patient’s symptoms. Dysmorphic facies may be diagnostic of a particular syndrome, especially in genetic disorders. For example, the face of patients with myotonic dystrophy type 1 has been described as shaped like a hatchet. Other neuromuscular conditions can be associated with organomegaly, endocrinopathy, scoliosis, or abnormalities of cardiac conduction, joints, or skin. Nerves may become enlarged (and may be palpable) in leprosy or in hereditary neuropathies (due to repeated demyelination and remyelination).
The neurological examination of the child with suspected neuromuscular disease naturally focuses on the PNS. Nevertheless, physicians should always include tests of the child’s mental status because in some neuromuscular disorders, changes in cognition accompany the PNS abnormalities. These cognitive changes are variable and can manifest as learning disorders or frank mental retardation.
Cranial nerves
Examination of the cranial nerves is described in detail in Chapter 1. Cranial nerves can be disrupted singly, in combination, or as part of a generalized polyneuropathy. Because most of the cranial nerves belong to the PNS, it is important to examine their function carefully in patients with suspected neuromuscular disease. In particular, the complaint of diplopia (double vision) often reflects weakness of eye movement (external ophthalmoparesis), which can result from damage to the oculomotor (CN III), trochlear (CN IV), and/or abducens (CN VI) nerves. Ptosis (drooping of the eyelid) uncommonly results from myopathy affecting muscles that elevate the eyelid; more often it is due to dysfunction of CN III or the sympathetic fibers that innervate those muscles or the NMJ that joins CN III to the muscles. Disturbance of facial sensation may reflect trigeminal nerve (CN V) dysfunction. Palsy of the facial nerve (CN VII) causes drooping of the face, such as in Bell’s palsy. Impaired auditory or vestibular function may suggest a lesion of the vestibulocochlear nerve (CN VIII). Difficulty speaking (dysarthria) and/or swallowing (dysphagia) can result from lesions affecting the glossopharyngeal (CN IX) or vagus (CN X) nerves. Neck flexion and turning and shoulder shrugging may be weak if the spinal accessory nerve (CN XI) is affected. With damage to the hypoglossal nerve (CN XII), the tongue may be weak or atrophic and fasciculations may be present.
When examining a patient, it is important for neurologists to remember that cranial nerve function depends not only on the cranial nerves themselves but also on related structures, such as the brainstem, NMJ, and cranial muscles. For example, external ophthalmoparesis can result not only from dysfunction of the oculomotor, trochlear, and/or abducens nerves but also from problems in the brainstem, NMJ, or extraocular muscles. Unilateral facial weakness usually signifies palsy of the facial nerve, but facial diparesis may result from neuropathy or myopathy.
Motor and gait
The motor examination includes assessment of muscle bulk, tone, and power. Atrophy—loss of muscle bulk—characterizes disorders of the LMN. For example, the sequelae of a severe Erb’s palsy may include atrophy of the deltoid and biceps muscles due to injury to C5 and C6 nerve roots. Likewise, hammer toes, flat feet (pes planus), or high-arched feet (pes cavus) from atrophy of the foot muscles may bespeak hereditary or other long-standing motor neuropathy, especially in older children and adolescents. Atrophy may also result from prolonged disuse. On the other hand, hypertrophy of muscles may result from overuse, which may be intentional (as in bodybuilding) or a consequence of repetitive involuntary movements such as dystonia (see Chapter 3). Some conditions cause pseudohypertrophy, in which muscle fibers are unchanged in size but fatty infiltration or increased connective tissue enlarge the muscle bulk. A classic example is the calves of boys with Duchenne muscular dystrophy (DMD), which become enlarged by fibrosis of the muscle. Other examples include glycogen storage diseases and endocrine disorders such as hypothyroidism and acromegaly.
Neurologists assess muscle tone, another element of the motor examination, by moving the patient’s body parts through their range of motion and feeling for resistance. Decreased tone (hypotonia) is most closely associated with lesions of the LMN, but it also can occur with cerebral or cerebellar pathology. Increased muscle tone in children usually occurs in one of two forms. Spasticity is increased resistance to passive movement that is more pronounced when the movement is made quickly. In contrast, the resistance of rigidity does not change with varying speeds of movement. Spasticity is typical of damage to the UMN in the brain or spinal cord, while rigidity is most characteristic of disorders of the extrapyramidal system, most commonly the basal ganglia.
An important component of the motor examination is the assessment of power. In older children and adolescents, neurologists usually use manual muscle testing, in which the examiner asks the patient to contract a muscle group and grades the strength of the contraction along a 0 to 5 scale (Table 18.2). Children younger than 6 years often are unable to cooperate with formal testing, but even preschoolers may cooperate with requests to walk (including walking on heels and on toes), rise from a sitting position, or perform a deep knee bend. An astute examiner will glean a great deal of information simply by observing the patient.
Watching a child walk gives valuable information about muscle power as well other neurological functions (including vision, balance, and coordination). By the age of 13 to 18 months, most toddlers can walk steadily with only occasional falls. At 18 months, most children can stand unassisted from a sitting position. By 24 months, most children have mastered the skills of running and jumping (often to their parents’ dismay). Table 18.3 lists some patterns of gait abnormalities commonly observed in children.
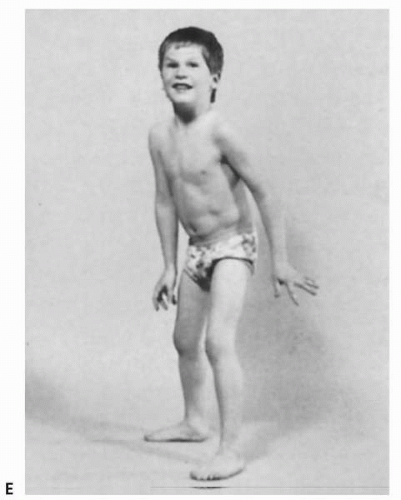
Recognizing the pattern of motor involvement as more prominent either proximally or distally is crucial in identifying its underlying cause. Children with proximal muscle weakness may have difficulty standing from a sitting position, and may need to use their hands to push on the floor and their knees, a maneuver known as Gowers sign (Fig. 18.1A-D,18.1E). A child with foot drop is likely to have trouble walking on his or her heels.
TABLE 18.2 MEDICAL RESEARCH COUNCIL SCALE FOR GRADING POWER DURING MANUAL MUSCLE TESTING
Grade 0
No contraction
Grade 1
Flicker or trace of contraction
Grade 2
Active movement, with gravity eliminated
Grade 3
Active movement against gravity
Grade 4
Active movement against gravity and resistance
Grade 5
Normal power
TABLE 18.3 SOME GAIT ABNORMALITIES SEEN IN CHILDREN
Type of Gait
Description
Possible Clinical Significance
Scissoring gait
Adduction of legs
Spasticity (often cerebral palsy or other spastic paralysis)
Weakness of hip muscles (especially thigh abductors); hip disease (e.g., Legg-Calve-Perthes disease, slipped capital femoral epiphysis)
Ataxic gait
Clumsy, poor coordination
Cerebellar disease or sensory abnormalities
Antalgic gait
Decreased weight-bearing on affected leg
Pain elicited by bearing weight
In general, muscle disease produces weakness and atrophy that is most pronounced proximally, while polyneuropathy results in predominantly distal abnormalities because it affects the longest nerves most severely. Exceptions include myotonic dystrophy, which is associated with distal weakness.
Sensation
Examination of sensation is highly subjective and requires the cooperation of the patient. Young children may be unable to provide useful information on formal sensory testing, although dramatically different reactions to similar stimulation in discrete body parts may reflect differential sensation.
Lesions of the anterior horn cell, NMJ, and muscle do not result in sensory changes. In contrast, nerve and nerve root damage, as well as CNS pathology, can cause abnormalities in sensation. A major goal of the sensory examination is to identify an anatomic pattern of abnormalities because, in analogy with motor abnormalities, sensory changes can be very helpful in localizing the site of the lesion.
Testing sensation of stimuli in a variety of sensory modalities may demonstrate dissociation of sensory changes, such that some modalities are affected more than (or to the exclusion of) others. Testing multiple modalities can be helpful in localization, for, as mentioned previously, separate tracts in the spinal cord carry information about pain and temperature (spinothalamic tracts) and about vibration and joint position (dorsal columns). Although neurologists sometimes test sensation of light touch, pin prick, cold, hot, vibration, and joint position, performing such an exhaustive examination is rarely practical in young patients. Rather, the examiner must selectively test those modalities most likely to elicit information relevant to the individual patient.
Deep tendon reflexes
Although disorders of the UMN typically produce hyperreflexia (overly brisk reflexes), diseases of the LMN tend to result in hyporeflexia or absent reflexes (areflexia). In length-dependent neuropathies, in which the longest nerves are preferentially damaged, a characteristic early sign is a bilaterally decreased Achilles tendon reflex. More focal reflex abnormalities occur in association with mononeuropathy, mononeuropathy multiplex, and radiculopathy Reflexes are largely spared in myopathies and NMJ disorders unless very severe weakness is present.
▪ FIGURE 18.1 Gowers sign is a classic finding in boys with Duchenne muscular dystrophy but may be present in children with proximal muscle weakness of any cause. In order to rise from a seated position on the floor, a child turns the body prone, then extends the arms and legs to raise the body (A). The hands are brought closer to the feet, in order to bring the center of gravity above the feet (B), and then one or both hands are used to push against the knees or thighs, enabling the child to straighten the back (C-E). (From Bickley LS, Szilagyi P. Bates’ Guide to Physical Examination and History Taking. 8th ed. Philadelphia: Lippincott Williams & Wilkins; 2003 with permission.)
▪ FIGURE 18.1(continued)
Coordination
In most cases of ataxia in children, a CNS process is responsible. Nevertheless, PNS abnormalities that impair sensation of joint position can also result in ataxia. Most of these conditions are neuropathies that affect sensory fibers, either exclusively or together with motor fibers. A few conditions directly afflict the cell bodies of the sensory nerves, but these are rare, especially in children.
LABORATORY TESTING
Although the history and examination remain the mainstay of diagnosis, neurologists use ancillary testing to aid in diagnosis. Serum levels of muscle enzymes rise in many muscle disorders, including inflammatory myopathies, Duchenne and Becker muscular dystrophy, and muscle injury. Creatine phosphokinase (CPK) is the enzyme most commonly measured, but levels of aldolase, lactic acid dehydrogenase, alanine transaminase, and aspartate transaminase may also be elevated.
An indispensable tool in the diagnosis of many neuromuscular disorders is electrophysiologic testing, which helps not only with localizing the affected area of the PNS but also with determining its severity and the underlying pathophysiology. Nerve conduction studies (NCS), one of two components of this testing, involve electrical stimulation of sensory and motor nerves and measurement of various parameters of the resulting action potentials. Because pathological processes disrupt these parameters in different ways, analysis of the pattern of abnormalities can aid in identifying the underlying disorder. Neuropathy is an example of a condition in which NCS results are often abnormal. Specialized techniques, such as repetitive nerve stimulation, are useful in diagnosis of NMJ disorders like MG.
Only gold members can continue reading. Log In or Register to continue