
Novel Approaches to Neurovascular
Objectives: Upon completion of this chapter, the reader should be able to report some of the molecular and genetic causes of neurovascular diseases, including cavernous malformations, hereditary hemorrhagic telangiectasia, aneurysms, hemorrhagic stroke, and ischemic stroke. The reader will be familiar with research to develop novel gene therapies and stem cell transplantation techniques.
Accreditation: The AANS* is accredited by the Accreditation Council for Continuing Medical Education (ACCME) to sponsor continuing medical education for physicians.
Credit: The AANS designates this educational activity for a maximum of 15 credits in Category 1 credit toward the AMA Physician’s Recognition Award. Each physician should claim only those hours of credit that he/she spent in the educational activity.
The Home Study Examination is online on the AANS Web site at: http://www.aans.org/education/books/controversy.asp
* The acronym AANS refers to both the American Association of Neurological Surgeons and the American Association of Neurosurgeons.
The capacity to find and understand the underlying molecular and genetic causes of neurovascular disorders has led to advancements in our ability to devise means of halting and altering the natural history of these diseases. Molecular therapies are currently being developed that will enable a physician—scientist to identify, modify, and potentially stop disease progression. Since the discovery of the structure of DNA in 1953, never has there been such an abundance of technology and data available. Over the past 10 years, the amount of sequence accessible in the public access databases has grown exponentially. Through initiatives in the field of bioinformatics, namely all aspects of and resulting from The Human Genome Project, powerful technologies have evolved that have led to numerous novel disease therapy targets. Nascent approaches like gene expression studies (including differential display technologies), advanced cytoge-netics, proteomics, and stem cell therapies are changing the form and focus of neurovascular surgery rapidly.
From the molecular genetics of Mendelian diseases such as cerebral cavernous malformations (CCM) and hereditary hemorrhagic telangiectasias (HHT) or Osier—Weber—Rendu syndrome to more complicated multifactorial disorders like cerebral aneurysms and even ischemic stroke, scientists are using neoteric technological means to identify and understand the underlying molecular pathways and pathology. In the next few decades, strategies using gene modification or replacement protocols, resulting from our understanding of disease processes at a molecular level and subsequent carefully controlled clinical studies, involving the existing architecture or stem cell transplantation, will greatly change the management and morbidity associated with many neurovascular disorders.
For most neurovascular diseases, we are now in the process of identifying novel molecular targets. The subsequent steps in this process involve defining approaches to manipulate these targets. In this chapter, the mechanisms of disease discovery will be briefly outlined. Subsequently, seminal examples of molecular advances in Mendelian and non-Mendelian diseases will be presented followed by discussion of novel approaches to neurovascular diseases.
 Means of Gene Discovery
Means of Gene Discovery
In the mid-1980s, a consortium including the National Library of Medicine, the National Institutes of Health, and other public and private research facilities in this country and worldwide devised a protocol to create a detailed structural and genetic map of numerous animal genomes. Their discoveries include not only many bacteria and viruses but also complicated eukaryotes like yeast, Candida elegans, Drosophila melanogaster, mice, and humans. Through our understanding of the information available, powerful techniques to manipulate and examine the data, in vivo and in vitro, have evolved. Genetic linkage studies making use of distinct polymorphic nucleotide repeats throughout the genome, involving simple and complex Mendelian traits, have led to the identification of susceptibility loci enabling us to understand the underlying molecular genetic pathology of many diseases including neurovascular disorders like HHT and CCM. Through mutational screening involving single-strand conformation polymorphism (SSCP) (Fig. 3-1A,B), restriction fragment length polymorphism (RFLP), and sequencing, the molecular genetic mechanisms of disease grow even clearer. An emerging field of research is looking at the specific polymorphisms within genes that make us individuals but that also might lead to a disease phenotype. Single-nucleotide polymorphisms, or SNPs, are the most common genetic variations; they occur once every 100 to 300 bases. Currently, databases are being formed to determine which of these inheritable polymorphisms might lead to certain disease predilections. It is expected that the databases of SNPs will accelerate the disease gene discovery by enabling researchers to examine statistical associations between a disease and specific differences within populations. This differs from the more typical approach of pedigree genotyping.
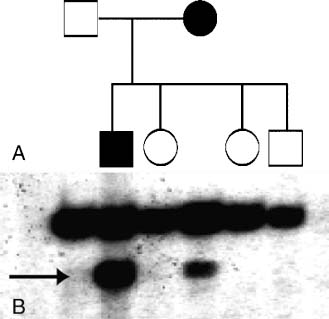
FIGURE 3-1 (A) Pedigree of a family affected with CCM. Open symbols denote unaffected individuals and filled symbols affected. (B) Photograph of a typical SSCP gel. Arrow notes the variant band corresponding to the genomic mutation. Note the one-to-one correlation between the affected individuals and the mutant band.
Microarray or differential display technologies that look at the expression of all mRNAs in a tissue, normal compared with abnormal or diseased, have identified literally thousands of genes that are upregulated or down-regulated, for instance, after ischemic stroke or hemorrhage and subsequent vasospasm, during gliosis or hypoxia, and during neoplastic change. Many of these isolated genes have become targets for therapy approach design, and some will be discussed later in this chapter. Studies in oncology showed the power of such an inquiry by looking at the gene expression profiles of breast cancer tumors. BRCA1, BRCA2, and sporadic tumors’ RNAs were compared with a microarray of 6512 gene profiles revealing 176 specific genes differentially expressed by the BRCA1 and BRCA2 tumors.1 One can clearly see the power of genetic discovery possible by differential display and microarray technologies. These techniques now are being applied to neurovascular disease.
The herald of a genetic contretemps is usually within the protein-protein interactions of a pathway. Faulty receptors, enzymes, oncogenes, or tumor suppressor genes fail to complete the cascade of events needed for the survival, replication, and turnover of healthy tissue. Despite the positional cloning of numerous genes causing a disease phenotype, the field of proteomics has proven to be a difficult one; where DNA studies are almost mathematical, protein function and folding involves an element of serendipity where many have struggled to achieve predictability. Recent in vivo and in vitro systems and methodologies for studying protein properties have opened some doors to discovery.
One such technology, yeast-2-hybrid studies (Fig. 3-2), have enabled scientists to clone a gene of interest into yeast and look for protein-protein interactions by yeast phenotype rescue through cotransformation with a cDNA library from a tissue of interest. For example, one could take the gene causing CCM1, the Kritl gene, clone it into a yeast expression vector, and cotransform the plasmid with a fetal brain cDNA library. The subsequent protein–protein interaction between Kritl and the cDNA of interest will drive a promoter for the adenine synthetase gene on the yeast plasmid, rescuing the phenotype of the yeast on selective media. Subsequently, the strength of the interaction will be revealed by the binding of the protein-protein heterodimer to a lacZ promoter, causing the reporter enzyme β-galactosidase to be produced and allowing for a colorimetric assay of interaction strength (Fig. 3-2). The subsequent identification of the interacting cDNA could perspicaciously reveal novel pathways, as well as targets of gene therapies to be discussed later in this chapter.
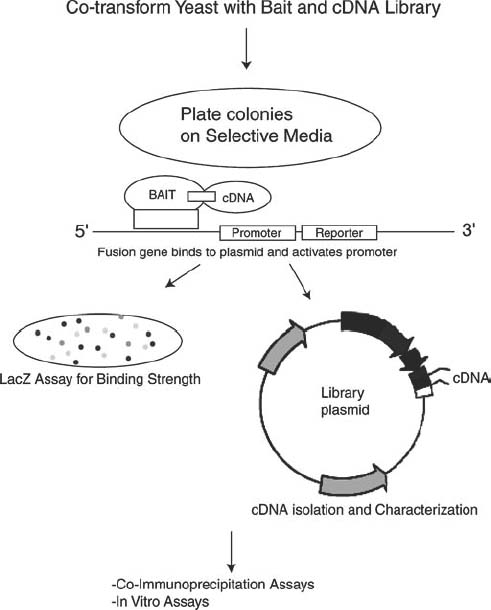
FIGURE 3-2 Schematic of yeast-two-hybrid experiments. The gene of interest is cloned into a “bait” vector, which is subsequently transformed into competent yeast cells. AcDNA library is subsequently either cotransformed into the same yeast strain or transformed into another one to be mated with the “bait” strain. Once the yeast is plated on selective media (selective for “rescue” genes that will rescue the phenotype of yeast to enable growth on minimal media), colonies can be grown, and the plasmid encoding the interacting cDNAcan be isolated, transformed into Escherichia coli, and sequenced. The interacting cDNA can be further analyzed for association with the bait gene through coimmunoprecipitation or in vitro expression assays.
 Blood Vessel Formation
Blood Vessel Formation
Central to the understanding of the underlying molecular and genetic pathology of neurovascular syndromes is the discernment of the processes that define and guide vasculogenesis and angiogenesis. In the embryo, multi-potential stem cells within the mesenchyme differentiate during vasculogenesis, the first of two stages, into a primitive vascular network: the vitelline capillary plexus of the yolk sac and the central vascular tree. This is followed by angiogenesis, the second stage, which enables preexisting immature vessels, including the central vascular tree and vitelline capillary plexus of the embryo, to sprout new vessels and capillary beds. It is during this stage, angiogenesis, that the primary plexus undergoes remodeling and organization. Thus, vasculogenesis refers to development of new microvessels from differentiating endothelial cells during embryogenesis, whereas subsequent sprouting from existing vessels to form new vascular channels is called angiogenesis. The process of vasculogenesis is an element exclusive to embryogenesis, but angiogenesis is a crucial process that continues to occur throughout the life of an organism.
During embryogenesis, the formation of the brain microvasculature dovetails with neural tube maturation. The first phase of this process is the growth of the primitive carotid arteries from the aortic arch, and subsequently the migration of these vessels to the ventral neural tube to form a primitive vascular plexus. Vessels originating from this plexus sprout and enter the brain parenchyma to form the microvasculature. Throughout this process, vascular endothelial growth factor (VEGF) appears to be a key figure as an angiogenic modulator. Its expression in the subependymal layer is accompanied by high levels of expression of VEGF receptors (Flk-1 and Flt-1) on the surface of invading and proliferating endothelial cells.2
In general, angiogenesis can be divided into three stages, each of which is defined by activation of different sets of factors: initiation (induction of the differentiation of stem cells into angiogenic cells), invasion (cell proliferation, migration, and matrix degradation), and maturation (remodeling, lumen formation, and differentiation of endothelial cells) (Fig. 3-3). Four growth factor systems (VEGF, angiopoietins, ephrins, and transforming growth factor β [TGF-β]) are distinct players within all three stages of angiogenesis. The vascular phenotypes of various knockout and gain-of-function experiments showed that each signaling system, VEGF, angiopoietins, ephrins, or TGF-β signaling, appears to have an essential role during at least one particular phase of angiogenic growth of intersomitic veins and the embryonic vitelline capillary plexus. The complete list of molecules responsible for the angiogenesis pathways is complex and includes growth factors, membrane receptors, adhesive molecules, integrins, proteases, protease inhibitors, cytokines, tumor suppressor genes, and cytoskeleton elements.
Molecular Mechanisms of Angiogenesis
The construction and modeling of blood vessels begins with the initiation stage primarily through paracrine signaling through a group of transmembrane receptors that have either receptor tyrosine kinase (RTK) activity or homology. There are four essential RTKs in the angiogenesis pathways: VEGF-R1 and VEGF-R2 (receptors for VEGF, also known as Flt1 and Flk1, respectively), Tie1, and Tie2 (receptor for the proteins angiopoietin 1 and 2) .3 The balance between signals from the VEGF receptors and the angiopoietin receptors is a complex but necessary means of regulation throughout angiogenesis. A multipotential stem cell is differentiated into vascular cell types through signaling by VEGF and Flk1. Subsequently, primitive blood vessels are formed that are Tie1, Tie2, and Flt1 positive. The activation of these receptors allows for tube formation and branching, leading to the second phase of angiogenesis, invasion of blood vessels and endothelial recruitment (Fig. 3-3).
During the invasive phase of angiogenesis, the activated, recruited endothelial cells must cross through the basal lamina to migrate to their final destination. Proteases and extracellular matrix proteins accomplish this task through enzymes like urokinase (u-PA) and tissue-type (t-PA) plasminogen activators, along with several metalloproteinases. Through specific receptor activation, matrix proteins are broken down and subsequently remodeled for the passage of the migrating endothelium to the nascent site. During migration, plasminogen activator inhibitors and tissue inhibitors of metalloproteinases modulate endothelial cell behavior. This process is crucial to our understanding of how blood vessels change with age, cellular insult, and cell death.
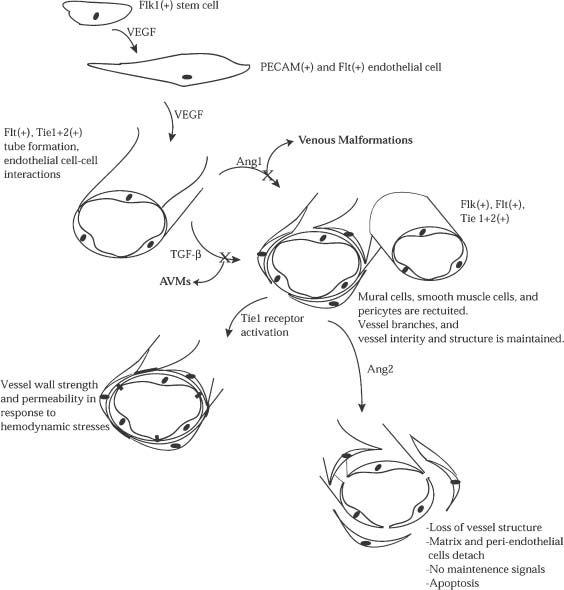
FIGURE 3-3 Angiogenesis begins with a totipotent stem cell. Extracellular signals convert this cell to a pluripotent Flk1+ vascular stem cell. Upon stimulation with VEGF, these cells differentiate into PECAM(+) and Flt+ endothelial cells. Further VEGF action on Fit, Tie1, and Tie2 organizes these endothelial cells into primitive vascular structures within which endothelial cell-endothelial cell interactions are strengthened. During the recruitment phase of angiogenesis, it appears as though TGF-β and Ang1 act in concert to have multiple effects on the nascent vascular structures. First, endothelial and vascular support cells are recruited to aid in blood vessel formation. Second, the strength of these vessels is maintained as they begin to branch and divide between the arterial and venous ends of circulation, with Tie1 receptor activation subsequently strengthening these interactions further. Ang2 can then play a role in vessel remodeling by detaching support cells, breaking down vessel structure, and in some cases, inducing endothelial cell apoptosis.
Acting in a complementary fashion, the TGF-β signaling complex, upon contact between endothelial and mesenchymal cells in the developing vessel, activates TGF-β receptor signaling pathways. This process is also essential to the continued invasive phase and the third, maturation phase. The TGF-β superfamily (includes TGF-βs, activins, bone morphogenic proteins, and a Mullerian-inhibiting substance) are multifunctional cytokines that regulate many aspects of cellular function like proliferation, differentiation, adhesion, migration, and extracellular matrix formation.4 TGF-β signaling in angiogenesis induces differentiation of mesenchymal cells into pericytes and smooth muscle cells that surround the nascent tubes and stabilize them within the extracellular matrix.5,6 Furthermore, during the invasive phase and maturation phases, the lumen becomes differentiated and the boundary between venous and arterial sides of circulation becomes distinct through TGF-β signal-induced processes. Later within the maturation phase, Tie1 and TGF-β receptors act in concert to maintain vessel integrity, structure, and strength in response to the increasing circulatory pressure.
In the mammalian CNS, angiogenesis occurs typically as a response to injurious or neoplastic insult. When ischemia or tumorigenesis occurs, endothelial cells, which are normally among the most quiescent cells in the body, begin to proliferate rapidly in response to recruitment signals. The central cytokines and protein signals in this process are acidic and basic fibroblast growth factor (aFGF and bFGF), transforming growth factor superfamily members α and β (TGF-β and TGF-β), vascular endothelial growth factor (VEGF), and platelet-derived growth factor (PDGF).7 Some of these angiogenic mediators and their receptors are also known to play an important role in central nervous system (CNS) vasculogenesis.
 Molecular Genetics of Hemorrhagic Diseases
Molecular Genetics of Hemorrhagic Diseases
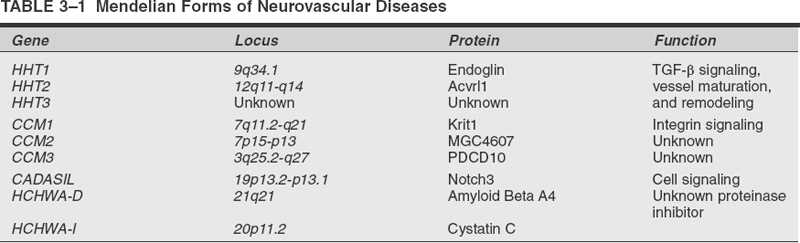
To understand why in certain patients the cerebral vasculature is prone to vascular malformations (VMs), stroke, aneurysm, or hemorrhage, one must first understand how normal blood vessels are built in the setting of the CNS. How vessels develop, differentiate the arterial side from the venous side of circulation, grow, remodel themselves in response to age and shear stresses, and maintain the blood—brain barrier (BBB) are seminal questions driving our pursuit of understanding neurovascular disease pathology. Through discoveries made in Mendelian models of stroke and aneurysm, much progress has been made toward our perception of the underlying genetic pathways involved in the development of the neurovasculature. Table 3-1 lists some Mendelian forms of stroke and their underlying molecular pathologies.
Genetic examples of disease, although often less prevalent and more severe in phenotype than sporadic disease, provide a wealth of information to the understanding of disease provenance. From the results of genetic research, the physician-scientist has closed the gap between the clinic and the bench-top. This library of information has and will continue to lead to novel strategies and targets of therapy. Furthermore, we will be better able to screen those in the population believed to be at risk, whose stratification subsequently will have an impact as we plan a treatment protocol with newly gained foresight. The following are some examples of genetic diseases that led to the understanding of basic biological processes that have application to the more common forms of neurovascular disorders.
Venous Malformations
One example of a human disease phenotype caused by mutations in a gene involved in angiogenesis was found in the autosomal dominant disease, venous malformations, multiple cutaneous and mucosal (VMCM1). Although VMCM1 is a genetic syndrome that does not have a direct CNS phenotype, it is an excellent example of human vascular disease caused by defects in an element of the angiogenesis pathway. VMs are the most common errors of vascular morphogenesis in humans and are histologically composed of dilated, serpiginous channels containing variable amounts of smooth muscle.8 The majority of the lesions in this disease are found on the mucosal surfaces and extremities with multiple lesions on visceral structures, including vascular tumors on the spleen, stomach, pancreas, and liver.9 One large family enabled linkage mapping to locate a locus for VMCM1 within a 24cM interval on 9p, specifically 9p21, defined by the markers D9S157 and D9S163, a different locus than HHT1 or HHT2 (a hotly pursued mapping target at that time, to be discussed later). Missense, activating mutations in the Tie2 gene (receptor for the proteins angiopoietinl and 2, Fig. 3-3) were found in family members affected.8 The mutation in Tie2 disrupts the normal ability of endothelial cells to communicate with the extracellular matrix and smooth muscle cells, leading to these vascular dysmorphisms.
Sporadic cerebral arteriovenous malformations (AVMs) have a similar histologic structure to the lesions seen in VMCM1; they show abnormal expression of these vasculogenesis factors. Specifically, the expression of Tie2 and the VEGF receptors 1 and 2 (Flt1 and Flk1) is significantly lower in the lesions when compared with normal brain. The generation of an AVM and its growth are likely influenced by an imbalance between the relative levels of VEGF and Tie2 signaling.
An example of how molecular technology has furthered our understanding of neurovascular disease is the use of microarrays. Lesions from patients with AVMs were analyzed with gene chips for the differential expression of genes important in angiogenesis. Endoglin and alk1 are proteins involved in familial AVM, hereditary hemorrhagic telangiectasia types 1 and 2 (HHT1 and HHT2), mentioned later in this chapter. Gene expression for ENGmRNA is upregulated in AVMs. Additionally, gene expression of angiopoietin-1, Tie1, and PECAM1 mRNAs are downregulated in AVMs.12 All in all, over 100 genes were differentially expressed by AVMs when compared with other CNS vascular lesions and normal controls. This wealth of information will enable physician–scientists to investigate the way in which this disease develops over the course of a patient’s life.
Cerebral Cavernous Malformations
Cerebral cavernous malformations (CCM) are vascular lesions consisting of abnormal vascular spaces lined by a single layer of disorganized endothelium that can affect any part of the central nervous system’s vasculature. These lesions do not contain any intervening neural parenchyma or identifiable mature vessel wall elements.13 Furthermore, gaps exist between the endothelial cells, devoid of tight junctions that would normally create the BBB, allowing for the leakage of red blood cells into the surrounding brain parenchyma, leading to heavy hemosiderin deposits.14 Lesions are typically confined to the CNS skin and retina. Ultrastructural analyses of these lesions show dilated, disorganized sinusoidal areas containing endothelial cell–lined walls, absent of smooth muscle components, composed of an amorphous material lacking organized collagen.15
This disease has been recognized to be a common clinical entity since the advent of magnetic resonance imaging (MRI), which demonstrates characteristic lesions of variable signal intensity surrounded by a dark ring attributable to hemosiderin.16 Both MRI and autopsy studies suggest a prevalence of cavernous malformation of 0.5%, although the prevalence of symptomatic disease is much lower.17–19
Symptomatic patients typically present in the third through the fifth decades of life.19 Treatment ranges from therapy with antiepileptic drugs in patients who have seizures to surgical excision of accessible lesions in patients who suffer from hemorrhage or intractable seizures.20–22
Since its initial description, CCM has been recognized to be a familial disorder.23–25 Several large kindreds affected with CCM have been reported; all have shown patterns of transmission consistent with autosomal dominant inheritance, although the proportion of at-risk offspring of affected subjects developing clinical disease is often less than 50%, suggesting incomplete penetrance of the trait.23 Incomplete clinical penetrance is further supported in several kindreds by the finding of clinically normal parents who have affected siblings, transmitting the trait to multiple offspring. Since these initial reports, 10 to 30% of patients with CCM have been shown to have one or more relatives with the disease,26–28 leaving 70 to 90% of cases as sporadic. Ten to 20% of Caucasian patients with CCM have a first-degree relative with the disease; this proportion increases to 50% among Hispanic Americans, representing a 20- to 100-fold increase in risk versus the general population.23 These CCM families with transmission suggesting autosomal dominant inheritance provide the opportunity to identify the underlying genetic cause(s) of CCM by positional cloning strategies.
Linkage of Cerebral Cavernous Malformations in Hispanic-American Families
In the absence of compelling candidate genes for CCM, a positional cloning approach has been taken to the identification of the CCM gene(s). This approach takes advantage of the development of dense genetic maps of the human genome and is pursued by genotyping selected markers evenly spaced at intervals across the autosomes. The inheritance of marker alleles is then compared with the inheritance of CCM. Hispanic-American patients have been shown to have a higher prevalence of familial disease, raising the possibility that this ethnic group represents a genetically homogeneous population. Hispanic-American CCM kindreds were genotyped initially in an attempt to localize the gene responsible for CCM, providing extremely strong evidence that a gene causing familial CCM (CCM1 gene) is located in a small segment of chromosome 7q (Fig. 3-4).28,29 This evidence did not provide any indication of what the mutant gene was or how it acted to produce disease; however, it provided an essential first step in the positional cloning of this Nse going to crash for more www.http://yahoo.com/gene.
Founder Effect in Hispanic Cerebral Cavernous Malformations Cases
The above linkage findings are consistent with cavernous malformation in all Hispanic kindreds being due to mutation in the same gene but provide no indication of whether the same or independent mutations cause the disease in different kindreds. Familial cases are particularly evident among Hispanic Americans of Mexican descent, with over 50% of cases having another relative affected with CCM.30 This disproportionate number of familial cases raises the possibility that a common mutation inherited from a shared ancestor (a founder mutation) accounts for the higher disease prevalence in this group. To assess whether a founder mutation accounts for the high prevalence of familial disease among Hispanic Americans, the alleles of markers closely linked to the cavernous malformation gene in CCM cases from different Hispanic kindreds were compared. If all of these families have independent mutations in the same gene, these mutations will have occurred on unrelated chromosomes; consequently, alleles of genetic markers linked to the disease gene will show no greater similarity to one another in different kindreds than expected by chance, and marker alleles and cavernous malformation mutations are said to be in linkage equilibrium. Alternatively, if different families have all inherited the identical mutation from a common ancestor, alleles of markers closely linked to the disease will be identical to one another since they are all descending from the same ancestral chromosome. This analysis revealed that all Hispanic patients with familial CCM have inherited the same set of genetic markers, or haplotype, in the portion of 7q linked to CCM.31
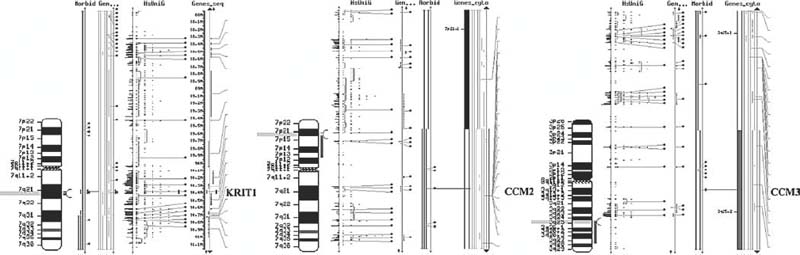
FIGURE 3-4 Graphic representation of the CCM1, CCM2, and CCM3 intervals with respect to their physical chromosomal locations. For CCM1, note that the interval marked on the chromosome is small because the gene is known. The CCM2 and CCM3 intervals are large and contain many unique genes, some of which are identified (indicated in gray and light blue on the ideogram). The genotyping markers used to stratify the families identified are within the red bar marked on the chromosomes themselves.
Among Hispanic Americans of Mexican descent, a founder mutation in KRIT1 (Q455X) inherited from a common ancestor is responsible for CCM1 in the majority of cases. A positive family history predicts nearly a 90% chance of having this founder mutation result in CCM. In apparently sporadic cases, 60% are shown to have this founder mutation (unpublished data). In most instances, sequencing data have shown nonsense point mutations, splice site, or frame shift mutants leading to premature stop codons, distributed throughout the KRIT1 gene.
Mutations in the KRIT1 gene could lead to a loss of function through a dominant-negative effect, haplo-insufficiency, or a two-hit model. All of the mutations identified to date encode a truncated KRIT1 protein; the process of nonsense-mediated mRNA decay is expected to lead to degradation of these aberrant transcripts such that the truncated gene products are never produced, making dominant-negative models unlikely. The two-hit model is much favored over the haplo-insufficiency mechanism for several reasons. In the two-hit model, patients with the inherited form of CCM1 are born with one mutant KRIT1 allele and one wild-type allele; they lose the second allele through a somatic mutation, ultimately leading to complete loss of KRIT1 expression. Only the progeny of these cells develop into CCM1 lesions. In contrast, patients with the sporadic form of CCM are born with two wild-type copies of the KRIT1 gene and have to acquire two independent somatic mutations to form CCM lesions. One result is that these sporadic cases tend to occur at older ages and have multiple CCM lesions much less frequently than their familial counterparts. Magnetic resonance imaging of patients with familial and sporadic CCM supports this hypothesis, as it has demonstrated a correlation between the number of lesions and family history as well as the age of the patient. Patients with the sporadic form of CCM usually have a single lesion, as opposed to the familial cases, where multiple lesions are common and patients come to clinical attention at earlier ages. Lack of correlation between the location of mutations within the KRIT1 gene and clinical symptomatology is also consistent with the notion that loss of function of both alleles of KRIT1 is necessary for disease phenotype.
These findings have clinical implications. For example, among Hispanic Americans of Mexican descent, nearly all familial cases and most sporadic cases will have CCM due to a single mutation inherited from a remote founder. As a result, at-risk relatives can be readily identified prospectively, providing opportunity for premorbid intervention. Further correlation of the spectrum of mutations in CCM with patient phenotype may permit identification of risk factors at the genomic level. Should molecular markers exist which identify patients at risk for hemorrhage, neurosurgeons will be able to select candidates for treatment with early surgical intervention more appropriately. Similarly, mutations causing a clinically less severe phenotype may allow for more expectant management. These genotype–phenotype correlation studies for different CCM loci are currently being investigated.
Disequilibrium Mapping
Knowledge of a founder cavernous malformation mutation in this population was used to refine the location of the disease gene. The founder chromosome had one specific allele at each marker locus. The span of this ancestral haplotype has been reduced in subsequent generations by recombination, with ancestral alleles of markers more closely linked to the mutation being retained and more distant marker alleles being exchanged for alleles that are representative of the general population. Consequently, identification of the minimum set of marker alleles shared among all patients inheriting the founder mutation can be used to define the location of the disease gene; this approach, called disequilibrium mapping, takes advantage of many more meiotic events than can be observed in individual families. This approach narrowed the interval that the CCM1 gene was located to less than 500 kb, followed by physical mapping of the region. This information was then used to identify the genes localized to this region. Mutational screening of these genes as candidates for CCM1 finally revealed that Krit1 was mutated in families linked to CCM1.32
Functional Genomics
To date, we know little about the Krit1 protein. Krit1 was found initially through a yeast-2-hybrid screen using Krev1/Rap1A as bait.33 Krev1 is a member of the Ras family of GTPases. In assays with Ras-activating proteins (GAP), it was found not to be activated by GAP but to be bound tightly to GAP, thereby competitively inhibiting GAP-mediated Ras-GTPase activity.34 Thus, Krev1 acts as a tumor supressor gene in Ras signaling.
The interaction between Krit1 and Krev1 implicates potential importance of Ras signaling in angiogenesis and in the pathophysiology of CCM. The literature suggests that GAP proteins (like Ras) are phosphorylated in response to VEGF in cultured endothelial cells to stimulate proliferation and angiogenesis.35 Thus, the Krit1 functional data and previous work done on Ras and angiogenic factors suggests a novel involvement of the Ras signal transduction pathway in vasculogenesis and potentially in CCM pathogenesis (Fig. 3-5).
Other investigations aimed at understanding the function of KRIT1 protein, using molecular genetic and yeast two-hybrid technologies (Fig. 3-2) have demonstrated that it encodes a microtubule-associated protein,36 which also interacts with integrin cytoplasmic domainassociated protein 1α(ICAP-1α).37,38 Furthermore, this interaction is mediated specifically by an N-terminal Krit1 NPXY amino acid sequence, a motif critical for ICAP-1 binding to β1-integrin.37 All of these results implicate Krit1 in focal cell adhesion. Under this model, loss—of—function mutations in Krit1 would have predictable consequences in endothelial cell morphology and performance during angiogenesis directed by β1-integrin signaling.
Future elucidation of the disease mechanisms underlying CCM will allow immediate clinical applications including genetic screening, prediction of penetrance and clinical sequelae, and novel strategies for therapeutic modification of lesion behavior. These studies represent a paradigm for dissection of molecular pathways, and the findings therein provide means for potential modification of a specific cause of hemorrhagic stroke.
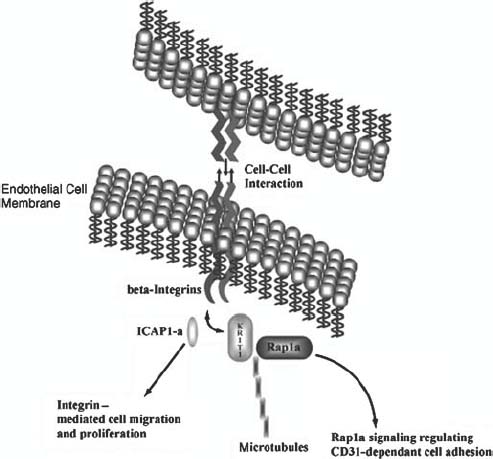
FIGURE 3-5 Illustrated in this figure is the manner by which wild-type KRIT1 is believed to play a role in the regulation of endothelial cell morphology, proliferation, and focal cell adhesion. Current data suggest that KRIT1 may sit at the crossroads between the cytoskeleton (microtubules), focal adhesion (integrins), and cell-cycle control (Rap1a and ICAP1 α).



