of obstructive apnea, reflecting an increase in inspiratory effort during the apnea (Fig. 18-3).
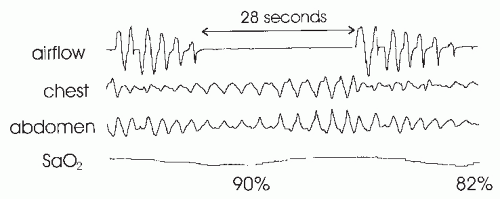 FIGURE 18-1 An obstructive apnea is characterized by absent airflow and persistent respiratory effort (movement of chest and abdominal bands). An arterial oxygen desaturation to 82% follows this 28-second apnea. The drop in the Sao2 to 90% is from a preceding apnea. In this case, the chest and abdominal tracing deflections progressively increase during the event. This is not always seen. |
different degrees of arterial oxygen desaturation. This could be relevant for many of the cardiovascular consequences of sleep apnea.
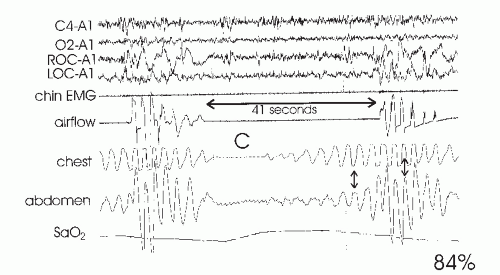 FIGURE 18-2 This tracing shows a 41-second mixed apnea. The initial portion (C) is a central apnea characterized by absent ventilatory effort. The small deflections in the abdomen tracing are from cardiac pulsations. After the central portion, an obstructive portion is noted. In this case, there is a progressive increase in chest and abdominal movements prior to apnea termination. In addition, there is paradoxical motion of the chest and abdomen tracings (small arrow), which vanishes after apnea termination. The apnea is followed by an arterial oxygen desaturation to 84%. In this case, an arousal is not seen in the central (C4-A1) or occipital (O2-A1) EEG tracings, although there is sudden movement in the eye tracings (ROC-A1 and LOC-A1). Here ROC and LOC are the right and left outer canthus electrodes, and A1 is the left mastoid electrode. |
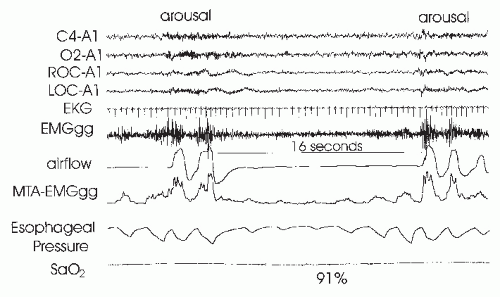 FIGURE 18-3 This tracing shows the end of one obstructive apnea followed by a 16-second obstructive apnea during NREM sleep. Arousal is noted at the termination of both events. The esophageal pressure swings increase toward the end of the event, consistent with a progressive increase in inspiratory effort. The moving time average MTA-EMGgg of the genioglossus EMG (EMGgg) was measured using a mouthpiece electrode. At the start of the apnea, the genioglossus activity falls; at apnea termination, there is a large increase in the EMGgg coincident with arousal and opening of the airway. |
could induce daytime sleepiness in the absence of arterial oxygen desaturation (14,15 and 16). These developments suggested that the sleep-disturbing consequences of respiratory events were as clinically important as the respiratory event-associated arterial oxygen desaturation.
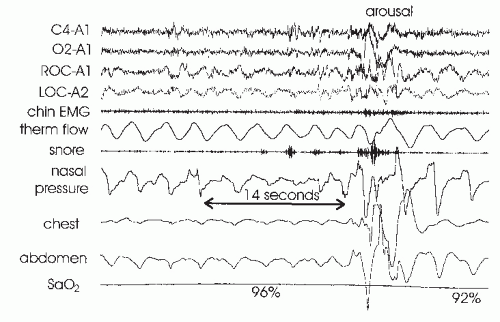 FIGURE 18-4 Obstructive hypopnea with arousal at event termination. Note that the decrease in airflow by nasal pressure is greater than by thermistor (thermal flow). Also note the flattening of the nasal pressure signal during the event. Obstructive hypopnea may be associated with paradoxical motion of the chest and abdomen band tracings, but this is not seen in this tracing. The reduction in airflow is associated with a 4% arterial oxygen desaturation and would therefore meet the hypopnea ciriteria based on oxygen desaturation. After arousal, the flattening in the nasal pressure signals resolves and the negative pressure swings decrease. |
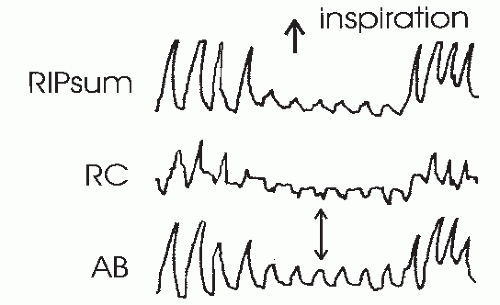 FIGURE 18-5 This tracing shows an obstructive hypopnea detected by RIP. Fluctuations in the RIPsum are estimates of tidal volume. The rib cage (RC) and abdominal (AB) band tracing are decreased during the hypopnea and also show paradoxical motion. |
is defined as the number of RERAs per hour of sleep. The RDI could then be used to assess the severity of SDB (24). The RAI can be defined as the number of arousals per hour of sleep associated with apnea, hypopnea, or a RERA.
TABLE 18-1 HYPOPNEA DEFINITIONSAa | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||
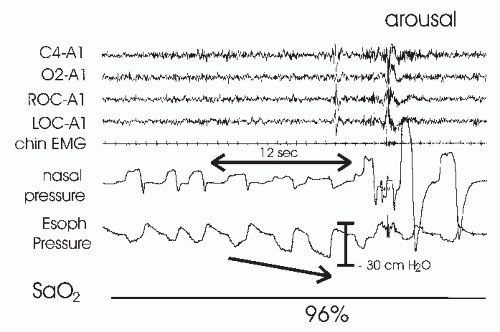 FIGURE 18-6 An RERA. The esophageal pressure shows a progressive increase in negative pressure followed by an arousal and reduction in the pressure swings. No desaturation was noted. There is also a reduction in flow by nasal pressure with a flattening in the inspiratory shape. After arousal, the flattening is abruptly reversed. Of note, this event would qualify as a hypopnea if an associated desaturation was not required (reduction in flow + arousal). |
The nasal pressure signal excursions (or those of the alternative hypopnea sensor) drop by >30% of baseline.
The event duration is at least 10 seconds.
There is a >4% oxygen desaturation from pre-event baseline.
At least 90% of the event’s duration must meet the amplitude reduction of criteria for hypopnea.
The nasal pressure signal excursions (or those of the alternative hypopnea sensor) drop by >50% of baseline
The duration of the event is at least 10 seconds.
There is a >3% oxygen desaturation from pre-event baseline or the event is associated with arousal.
At least 90% of the event’s duration must meet the amplitude reduction of criteria for hypopnea.
16-52), and the mean maximally negative esophageal pressure nadir was -37 cm H2O. There has been controversy as to whether the upper-airway resistance syndrome is a distinct entity or simply a milder form of OSAHS (29,30). Individuals without daytime sleepiness may have a RERA index of >10 per hour, although the mean RERA for a group of persons without symptoms is usually <10 per hour (31). Of note, the mean total arousal index in a group of normal persons using AASM criteria was 21 per hour in one study (32). The 95% confidence limit of normal for the arousal index was very wide due to high arousal rates in older patients. It is possible that respiratory arousals cause more potent sleep disruption than “spontaneous arousals.” However, this has never been experimentally addressed. In any case, there is some overlap in the arousal index between groups of normal subjects and patients with upper-airway resistance syndrome/mild OSAHS. If one uses nasal pressure monitoring for airflow and a definition of hypopnea that utilizes arousal, as well as a drop in the Sao2, most “upper-airway resistance” patients will have an AHI of >5 per hour and, hence, be classified as having the OSAHS. Alternatively, an RDI = AHI + RERA index will be >5 per hour if hypopneas are required to be associated with a 4% or greater desaturation.
imaging of the upper airway during wakefulness shows the smallest diameter at end expiration. Studies of the upper airway during general anesthesia (passive properties) have shown the upper airway of OSAHS patients to be narrower and more collapsible (49). When the Starling resistor model is applied to the upper airway, one can define a critical closing pressure (Pcrit) during sleep such that lower intraluminal pressures are associated with airway closure (50). In normal persons, Pcrit is negative, whereas in OSAHS patients, it is positive. That is, a positive intraluminal pressure is required to keep the airway open during sleep.
TABLE 18-2 FACTORS DETERMINING UPPER-AIRWAY PATENCY | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
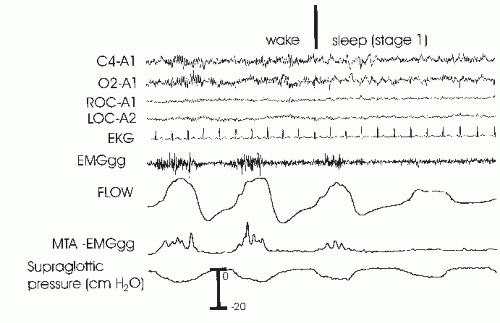 FIGURE 18-7 This tracing shows the onset of sleep with the coincident fall in genioglossus muscle activity and a fall in airflow with evidence of flow-limitation flattening. As the supraglottic pressure is similar but flow is lower, this means that upper-airway resistance has increased. |
and the upper airway opens. While it was once assumed that increasing genioglossus activity during obstructive apnea or hypopnea was driven by hypoxia and hypercapnia, a study of the effect of upper-airway local anesthesia suggests that mechanoreceptor stimulation (from increasingly negative pressure below the site of airway closure) is responsible for a large proportion of the augmentation (69). Traditionally a concept of a balance between negative inspiratory pressure tending to collapse the airway and upper-airway muscle dilating forces was assumed to determine the state of the airway. However, more recently the concept of passive collapse at sleep onset has gained favor. In fact, at sleep onset, the ventilatory drive decreases and supraglottic pressure actually may initially decrease (less negative) in some patients (although resistance increases). Upper-airway closure has also been documented during central apnea where there is no inspiration or negative collapsing forces (71). Therefore, suction pressure during obstruction may help keep the airway closed but is not necessary for the onset of airway occlusion.
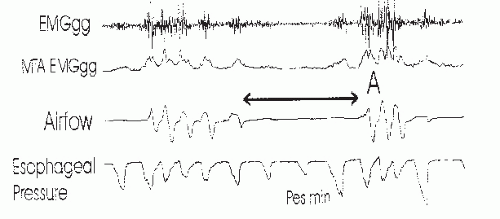 FIGURE 18-8 An obstructive apnea is shown with the raw genioglossus EMG (EMGgg) and the moving time average of the genioglossus EMG (MTA-EMGgg) as well as airflow and esophageal pressure. Note that the genioglossus EMG fall at airflow limitation (flattening) is noted on the last breath before apnea. At arousal (A), there is a large increase in genioglossus EMG. Note also that both EMGgg and the esophageal pressure deflections fall at apnea onset. In this case, the nadir in esophageal deflections (per minute) occurs on the second breath of apnea. |
does not occur until there is a preferential increase in upper-airway muscle activity (4). This is often associated with signs of cortical arousal, although the electroencephalography (EEG) changes may not meet AASM criteria (16), for example, the sudden onset of delta activity A recent study suggested that arousal can be detected at the termination of the majority of respiratory events if frontal electroencephalographs, as well as central EEG, is monitored (82). If cortical arousal does not occur, there is still believed to be a “state” change in the brainstem—socalled subcortical or autonomic arousals. The term autonomic arousal is used because an abrupt change in heart rate or blood pressure can be detected at apnea termination, even if cortical arousal is absent. While hypercapnia and hypoxia drive the increase in respiratory effort, the level of effort rather than individual values of hypoxia or hypercapnia seems to trigger arousal (13,83). Thus, the level of effort is an index of the combined arousal stimulus. Studies have suggested that information from upperairway mechanoreceptors may contribute to the arousal stimulus (84,85). In NREM sleep, arousal appears to occur when inspiratory effort reaches an “arousal threshold.” Normal subjects tend to arouse during mask occlusion when suction pressure reaches 20- to 40-cm H2O. In contrast, many patients with OSAHS arouse only after pressure reaches -60 to -80 cm H2O (13). The increased arousal threshold in OSAHS patients is probably due, in part, to chronic sleep deprivation or hypoxemia. Withdrawal of CPAP for even three nights has been shown to increase the arousal threshold in OSAHS. However, chronic CPAP treatment does not restore the arousal threshold to normal (86). Patients with OSAHS could have an intrinsically increased respiratory arousal threshold. Alternatively there could be damage to mechanoreceptors from years of snoring or to chemoreceptors from repetitive nightly stimulation. A study of respiratoryrelated evoked potentials (RREP) in patients with mild OSAHS suggested that there is a sleep-specific blunted cortical response to inspiratory occlusion (87). At least in these milder patients, there was no evidence of impaired mechanoreceptor function as the respiratory-related evoked potential was normal during wakefulness. One study found that the prolongation in event duration that occurs overnight in patients with OSAHS is secondary to a blunting of the cortical response as the level of inspiratory effort at apnea termination increased during the night (88). Another study found that the within-night variation in the arousal threshold followed the cycles of NREM sleep (89) with a higher arousal threshold associated with higher EEG delta power (deeper sleep).
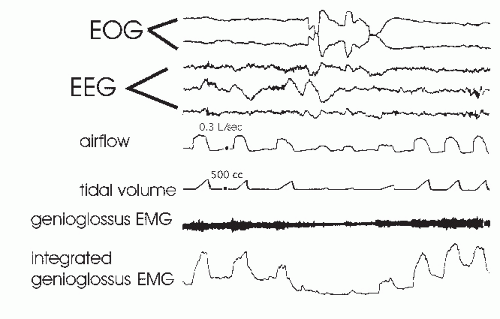 FIGURE 18-9 This tracing shows the fall in integrated genioglossus EMG activity and tidal volume during a burst of eye movements during REM sleep in a normal individual. The tracing illustrates that REM sleep is not homogeneous with respect to the effects on ventilation or upper-airway muscle control. (From Wiegand L, Zwillich CW, Wiegand D, et al. Changes in upper-airway muscle activation and ventilation during phasic REM sleep in normal men. J Appl Physiol. 1991;71:488-497, with permission.)
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|




