Fig. 1
Schematic overview of imputation. Imputation allows us to predict (‘impute’) genotypes which have not been directly genotyped on a microarray. In order to do so, microarray data is matched to a genome reference panel, which consists of densely genotyped (or sequenced) genomic data from multiple individuals. The genomic reference panels typically used for imputation are HapMap and 1000 Genomes. The most recent release of 1000 Genomes (phase I) provides a much larger set of SNPs (~37 million) relative to HapMap. This affords increased resolution of genomic regions. Imputation error increases as minor allele frequency decreases, as the imputation is based on an algorithm derived from a much smaller number of individuals (Marchini and Howie 2010). Accurate imputation of low-frequency SNPs requires genomic data from large numbers of individuals. HapMap 2 contains haplotype information on just 120 Europeans. However, HapMap 3 is based on data from 330 Europeans. Similarly, 1000 Genomes phase I includes far more haplotypes than 1000 Genomes pilot I, thus allowing for more accurate imputation of low frequency or rare SNPs. If rare variation is key to a particular trait, then 1000 Genomes Phase 1 (and later releases) may help identify these variants. Figure reproduced from Wood et al. (2013)
Consortia-based GWAS meta-analyses (typically imputed to HapMap 2) have identified a number of novel genomic regions associated with a range of smoking phenotypes, including smoking initiation and smoking cessation, as well as smoking quantity. Specifically, a nonsynonymous SNP (rs6265) in the brain-derived neurotrophic factor (BDNF) gene on chromosome 11 was found to be associated with smoking initiation. BDNF is involved in regulating synaptic plasticity and survival of cholinergic and dopaminergic neurons (Thorgeirsson et al. 2010; Tobacco and Genetics Consortium 2010). A variant near dopamine beta hydroxylase (DBH) gene on chromosome 9 was also identified in relation to smoking cessation (Thorgeirsson et al. 2010; Tobacco and Genetics Consortium 2010). DBH is an enzyme involved in the conversion of dopamine to noradrenaline. In relation to smoking quantity, genomic regions on chromosomes 8, 10 and 19 were identified, in addition to the 15q25 locus (Thorgeirsson et al. 2010; Tobacco and Genetics Consortium 2010). In particular, two genes with clear biological relevance were identified—two nicotinic receptor sub-unit genes (CHRNB3/CHRNA6) on chromosome 8 and CYP2A6 on chromosome 19 (Thorgeirsson et al. 2010). The CYP2A6 enzyme is principally responsible for the metabolism of nicotine (see chapter entitled Pharmacogenetics of Nicotine and Associated Smoking Behaviors; this volume). However, the proportion of phenotypic variance explained by these SNPs is far less than the variance explained by rs16969968-rs1051730 at 15q25, which explains ~1 % of the variation in smoking quantity.
High-density imputation has afforded additional benefits; for example, it has enabled high-resolution examination of the 15q25 region in relation to smoking quantity. Liu et al. (2010) used 1000 Genomes Pilot 1 data to fine-map the 15q25 region for association with smoking quantity. This allowed for analysis of virtually all common SNPs (MAF > 5 %) in the region, and offered a fivefold increase in marker density compared to HapMap 2. Using this imputation approach, combined with meta-analysis, Liu et al. (2010) identified rs55853698 as the variant with strongest evidence for association with smoking quantity at this locus. This variant, which is absent from HapMap 2, is located within a promoter region of CHRNA5, and is a plausible candidate for affecting mRNA transcription. However, this variant is also in very high LD with rs16969968. Conditioning on rs55853698 revealed a second independent signal at rs6495308, located within CHRNA3. Conditioning on both SNPs left no residual signal, suggesting that these two variants could explain the full signal at 15q25.1 in relation to smoking quantity (Liu et al. 2010).
In summary, GWAS has dramatically advanced our understanding of the genetic underpinnings of smoking initiation, cessation, and smoking quantity (see Table 1). Despite these successes, however, the total proportion of variance explained by all variants identified through GWAS to date is far less than the heritability estimates indicated by earlier twin studies. It is possible that rare variants (MAF < 1 %), which cannot be very accurately imputed using current genomic reference panels (given the limited numbers of haplotypes on which they are based) may account for this ‘missing heritability’. This is discussed in more detail in Text Box 1.
Table 1
Genomic loci identified through GWAS of smoking phenotypes
Author | Year | Phenotype | Chr | Gene | SNP | Notes |
|---|---|---|---|---|---|---|
Thorgeirsson | 2008 | Smoking quantity | 15 | CHRNA3 | rs1051730 | |
Liu | 2009 | Smoking status | 4 | near IL15 | rs4956302 | Marginal evidence for association (p = 8.8 × 10−8) in males only |
Furberg | 2010 | Smoking quantity | 15 | CHRNA3 | rs1051730 | Primary signal at this locus |
Smoking quantity | 15 | CHRNA5 | rs684513 | Independent signal (after conditioning on rs1051730) | ||
Smoking quantity | 10 | LOC100188947 | rs1329650 | |||
Smoking quantity | 10 | LOC100188947 | rs1028936 | |||
Smoking quantity | 19 | EGLN2 | rs3733829 | |||
Initiation | 11 | BDNF | rs6265 | |||
Cessation | 9 | DBH | rs3025343 | |||
Thorgeirsson | 2010 | Smoking quantity | 15 | CHRNA3 | rs1051730 | Primary signal at this locus |
Smoking quantity | 15 | near IREB2 | rs2869046 | Independent signal (after conditioning on rs1051730) | ||
Smoking quantity | 15 | AGPHD1 | rs2036534 | Independent signal (after conditioning on rs1051730) | ||
Smoking quantity | 19 | CYP2A6 | rs4105144 | |||
Smoking quantity | 8 | CHRNB3 | rs6474412 | SNP in shared LD block with CHRNA6 | ||
Liu | 2010 | Smoking quantity | 15 | CHRNA3 | rs1051730 | Original GWAS using HapMap (release 22) |
Smoking quantity | 15 | CHRNA5 | rs55853698 | 1000 Genomes (Pilot 1). SNP is in high LD with rs1051730 | ||
Smoking quantity | 15 | CHRNA3 | rs6495308 | Independent signal (after conditioning on rs55853698) | ||
David | 2012 | Smoking quantity | 15 | near CHRNA5 | rs2036527 | African American sample |
Text Box 1: The Missing Heritability Problem
Genome-wide association studies (GWAS) have been extremely successful in identifying genetic variants associated with a range of complex phenotypes. As we discuss, several loci associated with various tobacco use phenotypes have been identified, through large consortium-based efforts (Tobacco and Genetics Consortium 2010). This is in stark contrast to the candidate gene literature, where few reliable signals emerged after almost two decades of effort. Despite this, variants identified to date via GWAS explain less than half the heritability of complex phenotypes such as smoking behaviour estimated by twin and family studies. This is the so-called “missing heritability” problem (Manolio et al. 2009).
It is now clear that some missing heritability will be accounted for by variants that have not yet been identified via GWAS (Yang et al. 2010). This is in part because most common variant chips have relatively poor coverage in the minor allele frequency (MAF) spectrum below 5 %. Evolutionary theory predicts that mutations strongly affecting complex phenotypes will tend to occur at low allele frequencies (Visscher et al. 2012). There is, therefore, growing interest in the potential role of low frequency and rare variants (i.e., MAF <5 and <1 %, respectively). In other words, “missing heritability” is likely to be due to two factors (Visscher et al. 2012): (i) unidentified common variants with very small effects that cannot be detected by current GWAS using existing sample sizes, and (ii) rare variants not in sufficient linkage disequilibrium with variants on currently available genotyping chips and therefore undetectable via GWAS.
3 Phenotype Refinement
As we have discussed, GWAS approaches have proven highly successful in identifying genetic variants that are associated with a variety of tobacco use phenotypes (Thorgeirsson et al. 2010; Tobacco and Genetics Consortium 2010). For example, these approaches have confirmed the importance of the nicotinic receptor subunit gene cluster CHRNA5–A3–B4 in smoking quantity. Identification of novel genes implicated in tobacco use through genome-wide approaches (e.g., CHRNA5) has led to renewed interest in these genes and their products. However, these studies are limited in two important ways. First, they rely on self-report measures of smoking behaviour, often ascertained retrospectively and in different ways across different studies. This combination of self-report, retrospective recall, and the need for phenotype harmonisation, may introduce considerable measurement error. Second, these studies have not taught us what the fundamental mechanisms linking these genes to these behaviours are. Phenotype refinement provides a means by which to understand the specific mechanisms linking genetic variants to disease. Carefully designed and well-characterised phenotypes also offer greater measurement precision, in principle providing a ‘cleaner’ genetic signal, and improving the likelihood of effect replication. In this section, we discuss how phenotype refinement has advanced our understanding of these relationships.
3.1 In Vitro Studies and Animal Models
The rs16969968 variant in CHRNA5 is non-synonymous, resulting in an amino acid change (aspartate to asparagine) in the resultant α5 nicotinic receptor subunit protein. Research has shown that this variant is functional. In vitro studies have demonstrated that nicotinic receptor complexes containing the α5 receptor subunit featuring the aspartic acid variant exhibit a twofold greater maximal response to a nicotine agonist compared to α5 receptor complexes containing the asparagine variant (i.e., the risk variant robustly associated with heavier smoking) (Bierut et al. 2008).
‘Knockout’ mouse models have enabled us to further explore the impact of specific genes on specific phenotypes. A knockout mouse is simply a mouse in which a specific target gene has been selectively disabled. Studying the physiological characteristics or behaviours of such mice (e.g., assessing their response to nicotine), provides insight into the mechanisms which link the knocked out gene to disease. The development of α5 knockout mouse models has illustrated the role that the CHRNA5 gene plays in determining response to nicotine. Such mice are considered a model of individuals with reduced CHRNA5 gene function (i.e., carriers of the rs16969968 minor allele, associated with increased heaviness of smoking). Fowler et al. (2011) conducted an elegant series of experiments using the α5 knockout mouse model. Based on a self-administration paradigm, they observed that knockout mice responded far more vigorously than wild-type mice (i.e., mice with no inactivated genes) for nicotine infusions at high doses (see Fig. 2a). Whilst wild-type mice self-titrated delivery of nicotine dose to achieve a consistent, desired level during each test session (~1.5 mg kg-1), knockout mice did not, consuming greater amounts as dosage increased (see Fig. 2b). The authors proposed that deficient α5 signalling attenuates the negative effects of nicotine that normally serve to limit its intake, a conclusion which fits well with human research (i.e., smokers carrying the rs16969968 risk allele are likely to smoke more heavily than their counterparts without the risk allele). Fowler and colleagues further demonstrated that this effect could be ‘rescued’ in α5 knockout mice through injection of a lentivirus vector into the medial habenula (MHb), an area where α5 subunits are typically densely expressed, rescuing expression of α5 subunits in this region. The MHb projects to the interpeduncular nucleus (IPN). Notably, the pathway between the MHb and the IPN has previously been shown to regulate avoidance of noxious substances such as quinine (Donovick et al. 1970). Fowler and colleagues further observed reduced activity in the IPN in response to nicotine in knockout animals. Additionally, disruption of IPN activity was found to increase nicotine self-administration. However, both knockout and wild-type mice were indistinguishable in terms of responding for low doses of nicotine (see Fig. 2), indicative of shared experience of the rewarding, stimulatory effects of nicotine at low doses in both groups. These observations are complemented by a previous study by Jackson (2010), which also illustrated the initially shared and later differential experience of reward in response to an increasing dose of nicotine between α5 knockouts and wild-types, in this instance illustrated using a conditioned place preference task.
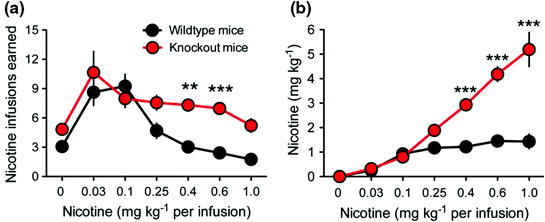
Fig. 2
 of Nicotine and Associated Smoking Behaviors
Receptors and Attention
Abstinence and Neurocognition: Implications for Cessation and Relapse
of Nicotine and Associated Smoking Behaviors
Receptors and Attention
Abstinence and Neurocognition: Implications for Cessation and Relapse
 Across Brain Regions and Neurotransmitter Interactions with Nicotinic Effects on Memory Function
Receptors, Memory, and Hippocampus
Across Brain Regions and Neurotransmitter Interactions with Nicotinic Effects on Memory Function
Receptors, Memory, and Hippocampus
 of Neuronal Nicotinic Receptors
of Neuronal Nicotinic Receptors




Self-administration of nicotine in α5 subunit knockout versus wild-type mice. Panel a Knockout and wild-type mice respond at an equivalent level for nicotine infusions at low doses. However, knockouts respond more vigorously than wild-types for high doses of nicotine. Panel b Wild-type mice self-titrate delivery of nicotine dose to achieve a consistent, desired level during test sessions (~1.5 mg kg−1), whereas knockout mice do not, consuming greater amounts as dosage increases. Figure reprinted by permission from Macmillan Publishers Ltd: Nature (Fowler et al. 2011), copyright 2011
Related posts:
of Nicotine and Associated Smoking Behaviors
Receptors and Attention
Abstinence and Neurocognition: Implications for Cessation and Relapse
Across Brain Regions and Neurotransmitter Interactions with Nicotinic Effects on Memory Function
Receptors, Memory, and Hippocampus
of Neuronal Nicotinic Receptors
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
