Fig. 1
TRPV4 inhibition does not alter the amplitude or frequency of spontaneous Ca2+ events in astrocyte endfeet after SAH. (a) Spontaneous Ca2+ oscillations recorded from 1.2 × 1.2-μm regions of interest placed on distinct astrocyte endfeet in a brain slice from one SAH animal in the absence (upper trace) and presence (lower trace) of the TRPV4 antagonist HC-067047 (10 μM). (b) Summary data of endfoot Ca2+ levels in the absence and presence of HC-067047 (10 μM) obtained from brain slices of SAH model rats. The term peak represents the average maximum Ca2+ concentration measured during individual spontaneous Ca2+ oscillations. (c) Summary data of the frequency of spontaneous Ca2+ oscillations ± HC-067047 (10 μM) obtained from 4-min recordings using brain slices from SAH model rats. For panels (b, c), recordings were made from 13 endfeet in four brain slices from four animals. NS, not statistically significant (P > 0.05), using paired Student’s t-test
TRPV4 Channels Do Not Mediate SAH-Induced Inversion of Neurovascular Coupling
To examine the impact of TRPV4 channels on SAH-induced inversion of neurovascular coupling, electrical field simulation (EFS) was used to induce neuronal action potentials in brain slices from SAH animals. Consistent with our previous studies [12, 13], EFS resulted in an increase in endfoot Ca2+ from a resting level of 169.6 ± 8.6 nM to a peak of 348.9 ± 33.0 nM (n = 5) (Fig. 2a, c). Associated with this EFS-induced increase in Ca2+, parenchymal arterioles encased by these endfeet constricted by 21.2 ± 4.3 % (Fig. 2a, b). This vasoconstriction in response to neuronal activation after SAH represents an inversion of the physiological vasodilation observed in brain slices from healthy control animals [8, 12, 25]. Incubation of brain slices with the TRPV4 channel antagonist, HC-067047 (10 μM), did not alter EFS-evoked increases in endfoot Ca2+ (peak Ca2+ after EFS: 330 ± 30.5 nM) or the magnitude of ensuing vasoconstriction (18.6 ± 3.1 % decrease in diameter) (Fig. 2a–c). These findings indicate that TRPV4 channels do not alter EFS-induced increases in endfoot Ca2+ or SAH-induced inversion of neurovascular coupling.
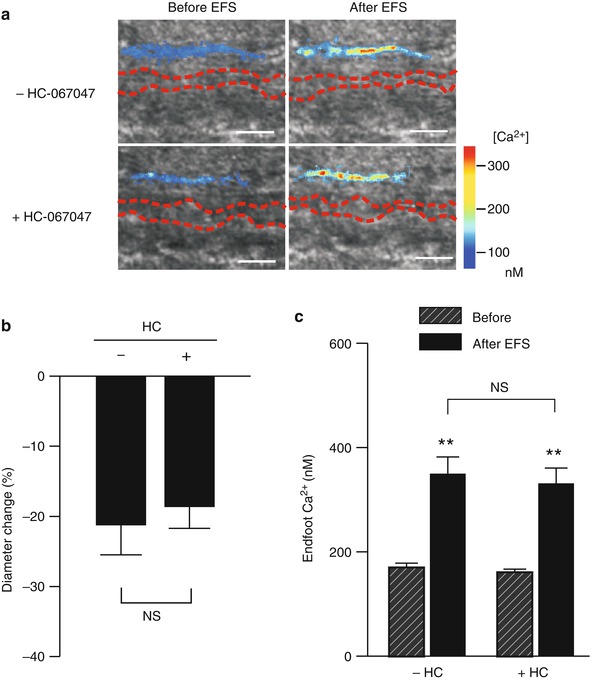
Fig. 2
TRPV4 channel inhibition does not influence SAH-induced inversion of neurovascular coupling. Parenchymal arteriolar diameter and astrocyte endfoot Ca2+ concentration were simultaneously measured using two-photon imaging and infrared–differential interference contrast (IR-DIC) microscopy. (a) IR-DIC images in the absence (upper panels) and in the presence (lower panels) of the TRPV4 antagonist HC-067047 (10 μM) obtained from brain slices of SAH model rats. Red dashes outline the intraluminal diameter of parenchymal arterioles using IR-DIC microscopy. Overlapping pseudocolor-mapped endfoot Ca2+ levels were obtained using the fluorescent Ca2+ indicator Fluo-4 and two-photon imaging. Scale bars, 10 μm. (b, c) Summary of EFS-evoked changes in arteriolar diameter (b) and astrocytic endfoot Ca2+ (c) obtained from SAH (n = 5 brain slices from three animals) model rats in the presence and absence of HC-067047. NS not statistically significant (P > 0.05), –HC-067047 vs + HC-067047, using paired Student’s t-test. **P < 0.001, before vs after EFS, using ANOVA followed by Tukey test
Discussion
Neurovascular coupling is an important physiological process enabling increased local blood flow to metabolically active regions of the brain. Recent evidence indicates that subarachnoid blood causes a fundamental change in NVC that could lead to pathological decreases in blood flow, rather than the “normal” physiological increases in blood flow associated with localized, task-dependent increases in neuronal activity. Altered Ca2+ signaling in astrocyte endfeet, in the form of high-amplitude spontaneous Ca2+ oscillations, are responsible for this SAH-induced inversion of neurovascular coupling. In this present study, we hypothesized that enhanced Ca2+ entry via TRPV4 channels contributes to the increased amplitude of spontaneous Ca2+ events observed in astrocyte endfeet after SAH. However, this does not appear to be the case, because pharmacological inhibition of TRPV4 channels did not alter endfoot Ca2+ signaling or the inversion of neurovascular coupling in brain slices from SAH model animals.
Astrocytes exhibit a diverse array of Ca2+ signaling events, including nerve-evoked propagating Ca2+ transients [8, 13, 25], spontaneous intracellular and intercellular propagating Ca2+ waves [7, 22], and nonpropagating spontaneous Ca2+ oscillations that can occur in either the cell body or in cell processes such as endfeet wrapping around intracerebral blood vessels [13, 17, 20]. Our recent evidence demonstrates that a marked elevation in the amplitude of spontaneous Ca2+ oscillations in endfeet rather than changes in nerve-evoked astrocyte Ca2+ signaling underlie inversion of neurovascular coupling after SAH [12]. In brain slices from SAH animals, high-amplitude Ca2+ oscillations in endfeet lead to increased K+ efflux into the perivascular space via increased activity of large-conductance Ca2+-activated K+ (BK) channels. This increase in basal extracellular K+ when summed with nerve-evoked astrocyte K+ efflux elevates extracellular K+ in the microenvironment surrounding parenchymal arterioles above the constriction threshold, leading to a polarity change in the neurovascular response from vasodilation to vasoconstriction.
In astrocytes from healthy animals, spontaneous Ca2+ oscillations reflect the release of Ca2+ stored in endoplasmic reticulum through activation of IP3-sensitive Ca2+ release channels (i.e., IP3 receptors) and occur independently from neuronal activity or mGluR activation [17, 20]. The activity of IP3 receptors is bimodally regulated by cytoplasmic Ca2+, with moderate increases in Ca2+ leading to an increase in IP3 receptor activation [9]. Thus, although requiring release of Ca2+ from intracellular stores, the amplitude of these spontaneous Ca2+ events in endfeet can be modulated by Ca2+ entering the cell through the plasma membrane. For example, Ca2+-permeable TRPV4 channels are present on astrocyte processes [3, 4], and activation of these channels caused an increase in the amplitude of spontaneous Ca2+ events in endfeet of brain slices prepared from healthy mice [6]. In addition, TRPV4 expression and function is upregulated in hippocampal astrocytes after cerebral ischemia [4]. However, our present data indicates that Ca2+ entry via TRPV4 channels does not contribute to the increase in amplitude of endfoot Ca2+ events occurring in SAH model animals. The present study does not exclude the possibility that Ca2+ entry through other TRP family members is upregulated in astrocytes after SAH. Although evidence suggests that normal native astrocytes do not express voltage-dependent Ca2+ channels, it is also possible that expression of these channels is upregulated in astrocytes after SAH. Alternatively, an increase in IP3 levels or other Ca2+-independent mechanisms leading to enhanced IP3 receptor activity may underlie the increase in the amplitude of spontaneous Ca2+ events in endfeet after SAH. Altered astrocyte Ca2+ signaling has been reported to occur in brain pathologies other than SAH, such as Alzheimer’s disease, ischemia/hypoxia, and epilepsy [2, 4, 5, 23]. Further, reactive astrogliosis and microglia activation have also been associated with multiple forms of brain injuries, including SAH [16, 21]. It has been postulated that Ca2+ oscillations in activated astrocytes may result in gliotransmitter release starting a cascade of events leading to excitotoxicity and brain damage [1]. Presently, the relationship between induction of reactive astrogliosis and the enhancement of astrocyte Ca2+ signaling is unclear and requires further investigation.
Related posts:
 Role of Erythropoietin in Aneurysmal Subarachnoid Haemorrhage: From Bench to Bedside
Modulation of Spreading Depolarizations
Care of Aneurysmal Subarachnoid Hemorrhage: State of the Art
Rate in the Sylvian Cistern Is Associated with the Severity of Cerebral Vasospasm After Subarachnoid Hemorrhage
Role of Erythropoietin in Aneurysmal Subarachnoid Haemorrhage: From Bench to Bedside
Modulation of Spreading Depolarizations
Care of Aneurysmal Subarachnoid Hemorrhage: State of the Art
Rate in the Sylvian Cistern Is Associated with the Severity of Cerebral Vasospasm After Subarachnoid Hemorrhage
 Changes in Brain Tissue Oxygen During Endovascular Treatment of Cerebral Vasospasm
Changes in Brain Tissue Oxygen During Endovascular Treatment of Cerebral Vasospasm
 Secondary Injury After Subarachnoid Hemorrhage in Light of Multimodal Advanced Neuroimaging, Intracranial and Transcranial Neuromonitoring: Beyond Vasospasm
Secondary Injury After Subarachnoid Hemorrhage in Light of Multimodal Advanced Neuroimaging, Intracranial and Transcranial Neuromonitoring: Beyond Vasospasm
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


