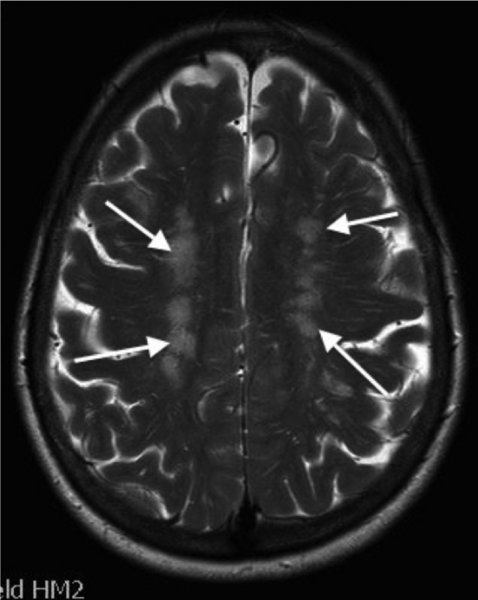
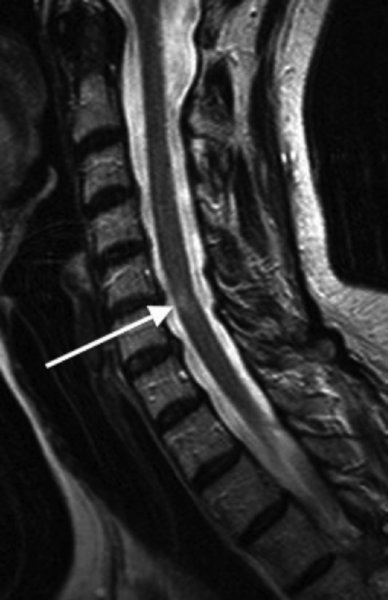
10 Marios Hadjivassiliou Academic Department of Neurosciences, Royal Hallamshire Hospital, Sheffield, UK Systemic lupus erythematosus (SLE) is a multi-system autoimmune disease of unknown aetiology with diverse clinical manifestations. The diversity of clinical presentations means that the diagnosis of SLE can be challenging. As such, patients may present to different medical specialties depending on their primary clinical complaint. This diversity is also reflected by the fact that the diagnosis of SLE relies on the presence of four or more of 11 criteria (Table 10.1), a mixture of clinical findings and laboratory abnormalities. The 11 criteria were originally devised in 1982 (Tan et al., 1982) to facilitate research into SLE, and these were then revised in 1997 (Hochberg, 1997). One of the criteria for diagnosis of SLE includes central nervous system (CNS) involvement. Table 10.1 The 11 SLE diagnostic criteria devised in 1982* *For the diagnosis of SLE, at least four criteria are required. In practice, patients clinically suspected of having SLE are usually tested for antinuclear antibody (ANA) and antibodies to double-stranded DNA (dsDNA) as the first approach in making a diagnosis. This is perhaps the reason why the Systemic Lupus Erythematosus International Collaborating Clinics group revised and validated the criteria in 2012 (Petri et al., 2012). Based on this revision, a patient can be diagnosed with SLE if they have kidney biopsy demonstrating lupus nephritis with positive ANA and dsDNA antibodies or if the patient satisfies four of the criteria (Table 10.1), assuming that these include at least one clinical and one immunological criteria (positive for ANA or anti-dsDNA antibodies). The prevalence of SLE is influenced by race and ethnicity with the highest rates reported amongst black and Hispanic populations in the United States (ranging between 40 and 100 per 100,000). The highest prevalences in the world have been reported in Italy, in Spain and amongst the Afro-Caribbean population in the United Kingdom (Danchenko et al., 2006). However, environmental trigger factors are equally important, as highlighted by the fact that SLE is rarely reported amongst the black population currently living in Africa. The female-to-male ratio is 11:1 during childbearing years, and the onset is usually after puberty (Manzi, 2001). The American College of Rheumatology published a nomenclature system for neuropsychiatric manifestations of SLE in an attempt to standardize and promote acceptance of the neuropsychiatric features of SLE (ACR Committee, 1999). This classification contained 19 neuropsychiatric conditions, including aseptic meningitis, cerebrovascular disease, demyelinating syndrome, headache, movement disorder (e.g. chorea), myelopathy, seizure disorder, cognitive dysfunction, mood disorder and psychosis for CNS involvement. Peripheral nervous system involvement includes acute inflammatory demyelinating polyradiculopathy, autonomic disorder, mononeuropathy multiplex, myasthenia gravis, cranial neuropathy, plexopathy and polyneuropathy (axonal sensorimotor neuropathy). Such classification has primarily been used for research purposes but may be less clinically helpful given that some of these ‘conditions’ represent symptoms (e.g. headache), others are based on case reports and are rare, and some are simply associations as a result of more than one autoimmune disease coexisting or coincidental in individuals, rather than aetiologically linked to SLE. For the purposes of this chapter, the neurological manifestations are divided into those affecting the CNS, including psychiatric manifestations; those affecting the peripheral nervous system; and those autoimmune neurological diseases that can be seen in SLE, probably as a result of more than one autoimmune disease coexisting in individuals. The prevalence of neuropsychiatric manifestations varies depending on what the spectrum includes; figures between 14 and 80% have been reported (Muscal and Brey, 2010). The term ‘encephalopathy’ has been used to describe diverse manifestations of CNS involvement in SLE, ranging from CNS vasculitis (usually presenting acutely with stroke-like episodes) to a slowly progressive CNS involvement, characterized by cognitive decline, depression, seizures, delirium and, rarely, psychosis. One of the commonest neurological complaints in patients with SLE is headache, a symptom that has been reported in up to 57% of patients. Headache is, however, a common complaint in the general population. A meta-analysis of all controlled studies on SLE and headache (eight in total) that included studies that reported the type of headache according to the International Headache Society classification, demonstrated that both migraine and tension-type headaches were present, but that the prevalence was not significantly different from that of the control populations studied. No particular pathogenic mechanism of headache in SLE was identified, and there was no association between the presence of headache and disease activity (Mitsikostas et al., 2004). Headache can be a feature of other CNS diseases affecting SLE patients, such as CNS vasculitis. Vasculitis of the CNS can be seen rarely in SLE and usually presents with headache, seizures, confusion and focal neurological deficits in the context of stroke-like episodes. Angiographic studies demonstrate typical arterial changes (irregularity of the arterial wall) in keeping with vasculitis, whilst magnetic resonance imaging (MRI) may demonstrate the presence of ischaemic changes affecting multiple vascular territories. This entity is different from what is seen in the context of anti-cardiolipin antibodies (often present in patients with SLE) where brain imaging demonstrates small patchy hyperintensities often attributed to cerebrovascular ischaemia (Figure 10.1). An MRI study of an SLE population demonstrated a prevalence of such lesions in up to 50% of patients. However, such abnormalities did not correlate with the presence of clinical CNS involvement, and the prevalence was the same in patients with SLE without neurological symptoms (Gonzalez-Crespo et al., 1995). Figure 10.1 MR imaging of the brain of a patient with antiphospholipid antibodies showing the characteristic ‘ischaemic’-looking abnormalities (arrows) primarily affecting the white matter. Another study used voxel-based morphometry to assess the regional grey matter volume in patients with SLE with and without neuropsychiatric manifestations. The study demonstrated reduction in the grey matter volume indicating atrophy affecting both groups of SLE patients, again unrelated to the clinical evidence of neuropsychiatric features. It therefore appears that a subclinical neurodegenerative and neuroinflammatory process affects a large number of patients with SLE (Cagnoli et al., 2012). CNS involvement in SLE is therefore common (probably as high as 50%) when assessed by imaging studies. In some cases, particularly those demonstrating increased white matter hyperintensities, CNS involvement may be related to the presence of anticardiolipin (aCL) antibodies and the prothrombotic tendencies associated with such antibodies. Such white matter involvement can be associated with cognitive deficits, and such deficits can be exacerbated by the presence of depression which is common in SLE. Chorea and ballismus (an extreme form of chorea) have been described in SLE and can be seen in about 1% of all SLE cases. Chorea as a presenting feature of SLE is even rarer. In a series of 51 cases of SLE, patients who developed chorea did so at the early stages of the disease, the chorea was transient and symmetrical and in 50% of cases it was associated with additional neurological manifestations (Bruyn and Padberg, 1984). Whilst chorea in SLE was initially thought to be secondary to ischaemic damage affecting the basal ganglia, more recent evidence suggests that this is not the case, given that the majority of cases have no imaging evidence of such ischaemia and that the chorea can improve with immunosuppressive treatment (Galanaud et al., 2000). Patients with SLE and chorea almost always have circulating anti-cardiolipin antibodies. The author has encountered a patient presenting with symmetrical ballismus and chorea, as a result of which the diagnosis of SLE was made. The patient had normal imaging and responded to steroids (prednisolone), but the chorea returned following reduction of steroid treatment. The patient responded to the introduction of mycophenolate, an immunosuppressant. These observations support an immunological rather than ischaemic aetiology for the chorea in SLE. Cerebral venous sinus thrombosis can be seen in SLE patients, usually as a result of the prothrombotic tendency in the context of anti-cardiolipin antibodies. It usually presents with headache and papilloedema and is treated with anticoagulants. There are case reports describing associations of SLE with diverse neurological and psychiatric diseases, including transverse myelitis, optic neuritis, cerebellar ataxia, multiple sclerosis (MS), idiopathic intracranial hypertension, anxiety, psychosis and others. There are some neurological diseases that seem to be associated with SLE, presumably on the basis of the coexistence of more than one autoimmune disease. However, some of these autoimmune diseases are over represented amongst patients with SLE and include myasthenia gravis, polymyositis and neuromyotonia. SLE is characterized by multi-organ microvascular inflammation and damage involving autoantibody production, immune complex formation and deposition and possibly intrathecal production of pro-inflammatory cytokines. SLE is therefore, above all, a disease of vessels. This explains the diversity of clinical presentations, including the involvement of the nervous system. Post-mortem studies in patients with CNS disease reveal a range of abnormalities that include, multifocal micro-infarcts as well as large infarcts, ischaemic demyelination and sometimes patchy demyelination reminiscent of MS. The microvasculopathy is suspected to arise from activation of complement and is probably the commonest histopathological brain finding in patients with SLE (Belmont et al., 1996). Many of the clinical manifestations are mediated by circulating immune complexes or are due to the direct effects of antibodies on cell surface antigens. Immune complex deposition may lead to complement activation and further localized inflammation. Blood–brain barrier damage is an important factor in the context of neurological manifestations of SLE. The two main candidate mechanisms for such damage invoke microthrombii in cerebral vessels, leading to ischaemia or immune-mediated activation of the endothelium leading to local cytokine production (Abbott et al., 2003). Antibodies against dsDNA, ribosomal-P proteins and N-methyl-D-aspartate (NMDA) receptor have been found in the cerebrospinal fluid (CSF) of patients with SLE and CNS involvement (Fragoso-Loyo et al., 2009). The same study examined the presence of IgG ANA, anti-dsDNA, anti-ribosomal-P, aCL, anti-beta-2 glycoprotein and anti-NMDA receptor antibodies as well as cytokines and chemokines in the serum and CSF of patients with SLE with neuropsychiatric symptoms. These assays were repeated 6 months later. There were no differences in the presence and levels of these antibodies, chemokines and cytokines between those patients with SLE and neurological involvement and those without. In both groups, the level of cytokines and chemokines decreased after 6 months, but this reached statistical significance in only the neurology group. The study did not identify a specific marker for neuropsychiatric manifestations. Up to 30% of patients with SLE have abnormal clotting times as a result of the presence of anti-phospholipid antibodies (also known as aCL antibodies and lupus anticoagulant). These antibodies (usually of IgG and IgM class) can predispose patients to recurrent miscarriage, thrombocytopenia, and venous and arterial thrombosis and can be linked to other neurological manifestations (e.g. chorea). In a series of 15 patients with SLE who had the highest aCL antibody titre amongst patients with SLE, six had a history of venous thrombosis, five had cerebral infarcts, five had thrombocytopenia and two each had pulmonary hypertension and multiple abortions (Harris et al., 1983). The presence of repeatedly positive aCL antibodies in patients with SLE has also been associated with cognitive dysfunction (Muscal and Brey, 2010). A similar association has been observed in patients with SLE who have anti-glutamate receptor antibodies. The mechanism of such an association remains obscure, particularly given that such patients may have entirely normal brain imaging. Primary Sjögren’s syndrome (PSS) is one of the commonest autoimmune diseases. It is characterized by lymphocytic infiltration of the exocrine glands leading to enlargement of the glands and clinically manifesting with dry mouth (xerostomia) and dry eyes (xerophthalmia). Secondary Sjögren’s syndrome occurs in association with other connective tissue diseases such as rheumatoid arthritis, SLE and scleroderma. PSS can be associated with other organ involvement, including lungs (pneumonitis), renal involvement, pancreatitis, myositis and occasionally lymphoma (Mori et al., 2005). The criteria for diagnosis of PSS have been the subject of a number of workshops, most recently in 2012 at a meeting of the Sjögren’s International Collaborative Clinical Alliance Research Group. The diagnostic criteria were approved by the American College of Rheumatology (Shiboski et al., 2012). The diagnosis relies on the presence of two out of three of the following: (i) positive serum antibodies known to be associated with PSS (anti-Ro and anti-La), (ii) demonstration of xerophthalmia using a special ocular-staining score and (iii) labial salivary gland biopsy showing focal lymphocytic sialadenitis. PSS affects up to 4% of the adult population, a figure that makes it one of the three most common autoimmune diseases. In PSS there is a female-to-male ratio of 9:1. The onset of the disease is usually in the fourth or fifth decade of life, but it can affect younger individuals. Point prevalence studies, however, have shown that PSS is approximately seven times higher in the elderly population aged 71–74 years when compared to individuals aged 40–44 years (Haugen et al., 2008). PSS is associated with an increased risk of non-Hodgkin’s lymphoma. A recent study from Norway demonstrated that the risk of Hodgkin’s lymphoma in patients with PSS is increased nine times when compared to that in the general population (Johnsen et al., 2012). The interest in neurological manifestations of PSS started in the 1980s following the publication of a series of papers by a group of researchers based at John Hopkins Hospital, Baltimore, United States (Alexander et al., 1981). This same group came up with a figure of prevalence for neurological involvement of 20%. Prevalence figures for CNS involvement in PSS remains a controversial issue, reflected by the very wide range reported in different studies (from 1 to 100%). Possible contributory factors to the controversy include the type of diagnostic criteria used for PSS, the inclusion of patients with secondary SS (secondary SS can be seen in the context of other connective tissue diseases such as SLE, rheumatoid arthritis etc.), the inclusion of neurological diagnoses that have no aetiological link to PSS, geographical variations and referral bias (Soliotis et al., 2004). As PSS is a common disease often associated with other autoimmune diseases, the coexistence of PSS with common autoimmune neurological diseases such as MS has to also be considered. The concept that PSS may mimic MS was first put forward by the same Baltimore group in 1986 (Alexander et al., 1986). The authors described a range of neurological signs in patients with PSS, including optic neuritis, intranuclear ophthalmoplegia, cerebellar ataxia and pyramidal weakness. In some cases, the neurological involvement followed a relapsing-remitting course, a pattern that is typical of MS. Subsequent studies, however, failed to identify an increased prevalence of PSS amongst patients with relapsing-remitting MS. The overall conclusion of such investigations and publications was that, rarely, PSS can be associated with MS-like features. Such features may follow a progressive course with sequential multifocal brain involvement, including spinal cord inflammation in the form of transverse myelitis (Figure 10.2) and optic nerve inflammation in the form of optic neuritis. It is rare for PSS patients to have brain MRI findings that are indistinguishable from MS unless the two diseases coexist. Figure 10.2 Inflammatory cord lesion (arrow) in a patient with myelopathy due to primary Sjögren’s syndrome. CNS vasculitis can complicate PSS in the same way that it can be seen in the context of any connective tissue disease. The diagnosis is based on brain biopsy following typical angiographic changes on cerebral angiogram. PSS patients with CNS vasculitis present like those with any other CNS vasculitis with an encephalopathy, which is often associated with seizures and focal neurological deficits. Patients respond to immunosuppression with steroids and cyclophosphamide. Widespread myoclonus can be a very prominent feature in PSS and responds to the use of clonazepam and other anticonvulsants. The aetiology of this remains obscure. By far, the most common and better characterized form of peripheral nerve involvement in PSS is that of sensory ganglionopathy. This is a form of asymmetrical, purely sensory peripheral nerve involvement that affects the dorsal root ganglia. It is often associated with sensory ataxia and can often be the presenting feature of PSS. In a series of 92 patients with PSS-associated neuropathy, 93% were diagnosed with PSS after neuropathic symptoms appeared (Mori et al., 2005). The commonest form of peripheral neuropathy was sensory neuronopathy (59%), with mononeuropathy multiplex being the second but much less common type (12%). Sensory ganglionopathy in PSS is slowly progressive but ultimately disabling because of the severe sensory ataxia. There are no published large treatment trials, and because of the nature of the slow progression, such patients are usually under observation without any active treatment being considered. Immunotherapies have been used in small uncontrolled and retrospective cases using intravenous immunoglobulins, steroids and cyclophosphamide. One retrospective study found that up to 18% of patients with PSS and sensory ganglionopathy improved with the use of steroids (Mori et al., 2005). The common presence of secondary SS in patients with SLE led to the suggestion that the two diseases may share common pathogenetic features. SLE can be considered as a disease with several clinical subgroups each characterized by particular autoantibodies. These two diseases share such autoantibodies (e.g. anti-Ro and anti-La antibodies), and some clinicians consider PSS as a subset of SLE, characterized by homing receptors that allow lymphocytic infiltrates into particular sites such as the lacrimal and salivary glands (Fox and Liu, 2006). A potential role for anti-Ro antibodies in the pathogenesis of neurological involvement comes from in vitro studies where serum from patients with PSS containing anti-Ro antibodies was shown to stain the cytoplasm and cell membranes of endothelial cells derived from umbilical vein and from brain tissue (Alexander et al., 1994). Sensory ganglion cell destruction associated with lymphocytic infiltration has been seen in cases of PSS and sensory ganglionopathy on dorsal root ganglion biopsy. PM examination findings included diminution of sensory ganglion neurones as well as loss of nerve fibres in the dorsal columns of the spinal cord. Mild lymphocytic infiltration was also noted. The overall picture was suggestive of a ganglioneuritis affecting the sensory neurones (Mori et al., 2005). Sensory ganglionopathy is also a common manifestation of other autoimmune-mediated disorders such as paraneoplastic neurological syndromes, particularly those associated with anti-Hu antibodies (discussed further in this chapter) and gluten-related neurological dysfunction. Another pathophysiological mechanism seen in the context of PSS is that of small vessel vasculitis that can less commonly complicate PSS but almost certainly accounts for some of the neurological manifestations, in particular mononeuropathy multiplex and CNS vasculitis.
Other Autoimmune Disorders: Systemic Lupus Erythematosus, Primary Sjögren’s Syndrome, Gluten-related Neurological Dysfunction and Paraneoplastic Neurological Syndromes
Systemic lupus erythematosus
Introduction
Epidemiology
Neurological manifestations
CNS manifestations
%Encephalopathy
%Chorea
Other neurological manifestations and associations
Pathophysiology of neurological manifestations
Primary Sjögren’s syndrome
Introduction
Epidemiology
Neurological manifestations
Pathophysiology of neurological manifestations
Gluten-related neurological dysfunction
Introduction
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


