The complexity of clinicopathologic relationships along the frontotemporal lobar degeneration (FTLD) spectrum. The major clinical syndromes along the FTLD spectrum are shown in the top row of the slide, and the pathologic diseases are shown below. The width of the colored bar in each clinical syndrome represents the approximate percentage of that syndrome that has been associated with each color-coded pathology below. All of the pathologies under the Frontotemporal lobar degeneration (FTLD) heading are described in detail in Chapter 13. A portion of most of the clinical syndromes may be attributed to Alzheimer’s disease pathology (shown in white). For example, the majority of cases of svPPA are associated with FTLD-TDP43 type C pathology (lavender), while the majority of PSPS cases are due to PSP 4R tauopathy (salmon). Abbreviations: bvFTD = behavioral variant frontotemporal dementia, svPPA = semantic variant primary progressive aphasia, nfvPPA = non-fluent variant PPA, FTD-MND = FTD-motor neuron disease, CBS = corticobasal syndrome, PSPS = progressive supranuclear palsy syndrome, FTLD-tau = tau-positive FTLD, FTLD-TDP = FTLD with TDP-43 (transactive response DNA-binding protein 43) proteinopathy, FTLD-FUS = FTLD with fused in sarcoma accumulation, CBD = corticobasal degeneration, PSP = progressive supranuclear palsy, aFTLD-U = atypical FTLD with ubiquitin inclusion, BIBD = basophilic inclusion body disease, FTDP-17 = frontotemporal dementia with parkinsonism-17, CTE = chronic traumatic encephalopathy, AGD = argyrophilic grain disease, MST = multisystem tauopathy, GGT = globular glial tauopathy, NIFID = neuronal intermediate filament inclusion disease, NIBD = neurofilament inclusion body disease, NOS = not otherwise specified.
For most of the twentieth century, progressive dementias in which deteriorating judgment and conduct were the most salient features were diagnosed “Pick’s disease.” The histopathologic diversity of FTLD was highlighted in 1974 (Constantinidis et al., 1974), and followed soon afterwards by Elizabeth Warrington’s seminal description of focal word and object agnosia (Warrington, 1975), which culminated in the description of SD in 1989 (Snowden et al., 1989). Starting in 1982 with a clinicopathologic report of six cases, Marsel Mesulam reinvigorated interest in PPA (Mesulam, 1982). Since the 1980s, FTD has been captured in formal diagnostic criteria, beginning with descriptive studies from the Lund (Gustafson, 1987) and Manchester (Neary et al., 1988) groups that renewed interest in the behavioral phenotype of FTD and culminated in clinical and pathologic diagnostic criteria (Brun et al., 1994). These were followed several years later by more formalized clinical rules specifying three subtypes characterized by disruption of conduct, temperament, and socialization; progressive non-fluent aphasia; or progressive semmantic aphasia and associative agnosia (SD) (Neary et al., 1998). These diagnostic criteria presented rigid core and peripheral criteria for the syndromes, and thus were rather complex. Simpler criteria proposed in 2001 (McKhann et al., 2001) emphasized ease of use, by stipulating that FTLD is characterized by early and progressive changes in personality or language of sufficient severity to cause functional disability. These criteria did not gain wide acceptance, owing to a perception of operational vagueness. The contemporary criteria, developed in 2011 for the behavioral (Rascovsky et al., 2011) and aphasia (Gorno-Tempini et al., 2011) phenotypes, have brought flexible application (which accommodates the diversity of presentations), established a hierarchy of diagnostic confidence (i.e.,“possible,” “probable,” or “definite” bvFTD), and also incorporated neuroimaging features, as well as genetic and neuropathologic typing into the diagnostic process. These latest criteria have been incorporated, in simplified form, into DSM-5 (American Psychiatric Association, 2013a).
Demographic distribution of FTLD is an important consideration in the clinical assessment. FTLD usually manifests in midlife, i.e., in patients who are 45–65 years old, although cases developing at ages as young as 21 (Snowden et al., 2004) and as old as 85 (Gislason et al., 2003) have been reported. The majority of cases in many bvFTD case series are male, whereas the aphasia phenotypes appear to have an even sex distribution. Age at presentation may also influence the presenting phenotype (Velakoulis et al., 2009), with younger cases showing a more “psychiatric” presentation, and elders a mainly “cognitive” presentation. Epidemiologic studies have also proposed that age-related phenotypic variation is a cause of under-ascertainment of cases (Ibach et al., 2003), noting that FTLD may be more common than is recognized among the elderly (Knopman et al., 2004; Borroni et al., 2010) where amnesic presentations are frequently seen and mistaken for Alzheimer’s disease (AD) (Baborie et al., 2012; Onyike et al., 2013).
Clinical characteristics of FTLD spectrum diseases
FTLD phenotypes are, as noted in earlier chapters, largely determined by the distribution of neurodegeneration – i.e., by particular patterns of frontotemporal or frontostriatal circuit dysfunction as a result of degenerative pathology. The regional patterns are, at least in some cases, related in part to the underlying histopathologic types (Rohrer et al., 2010; Whitwell et al. 2012a). Symptoms referable to extrapyramidal, brainstem, and upper/lower motor neuron systems are commonly seen because degeneration affects these areas in many cases. Ultimately phenotypes are stereotypical in most respects, being correlated with typical atrophy patterns (seen as brain imaging signatures) that may be linked at least probabilistically to histopathologic types. This forms the basis for differential diagnosis, such that a case of bvFTD or PPA with accompanying parkinsonian symptoms will be differentiated from idiopathic Parkinson’s disease (PD) because the clinical features (behavioral and/or language) will extend well beyond and dominate the clinical phenotype. Furthermore, the patient may manifest motor features that are not part of typical parkinsonism, such as gait apraxia. That is, a wide range of clinical phenomena can accompany the typical features of FTLD syndromes. The clinician should not be discouraged by this complexity; we have learned that as experience with the FTLD syndromes grows, one’s diagnostic behavior shifts imperceptibly from a synthetic to a (faster) pattern recognition process. At the same time, ambiguous cases are not uncommon so it is important to have a firm grounding in the symptoms and signs that signal FTLD, the laboratory data that confirm or discount the diagnosis, and the less formal “rules of thumb,” “red flags,” and caveats that may help in resolving ambiguous cases. We will first briefly touch on the major FTLD clinical syndromes and then discuss their differential diagnosis.
Major FTLD clinical syndromes
bvFTD appears to be the most common variant of FTLD. Its essence is a disruption of conduct, temperament, judgment, self-control, and socialization, with heterogeneity in symptom combinations according to the distribution of degeneration in pertinent brain systems. The typical features and their variations are reviewed in Chapters 2, 3, and 4.
The progressive aphasia phenotypes – non-fluent/agrammatic, semantic, and logopenic – feature focal disruptions of language in symptom combinations that are discussed in detail in Chapter 5.
Many investigators increasingly view conditions with prominent motor symptoms as falling within the FTLD spectrum. These include PSP and CBS, reviewed in detail in Chapter 7, and ALS, reviewed in detail in Chapter 6.
Other clinical phenotypes
Attention has been drawn to a variety of uncommon clinical phenotypes associated with FTLD spectrum pathology or with causal mutations. These include atypical “neurologic” presentations, such as posterior cortical atrophy or AD-like presentations. It is becoming increasingly recognized that FTD patients with GRN mutations have more parietal involvement than typical FTD patients (Masellis et al., 2006; Whitwell et al., 2007, 2012b; Le Ber et al., 2008). In some cases, this is an early presenting feature and is associated with prominent early limb apraxia or visuospatial dysfunction. We have seen one case of FTLD-TDP type A due to a GRN mutation that presented as a classic case of posterior cortical atrophy with lateralized visual impairment and topographic disorientation. It is also clear that, despite the exclusion criterion of episodic memory impairment in the new diagnostic criteria for both bvFTD and PPA (which is important from a research perspective), some patients with FTD exhibit prominent episodic memory impairment (Hornberger et al., 2010, 2012). Thus, clinicians should keep in mind that not every patient with PPA or bvFTD will have preserved episodic memory and visuospatial function.
Atypical “psychiatric” presentations of FTLD are seen, usually featuring states such as mania, depression, obsessive–compulsive disorder (OCD), attention-deficit hyperactivity disorder (ADHD), and psychosis. Another entity, designated “FTD phenocopy,” has been described. These are cases who present with a full complement of symptoms and signs, and an overall clinical appearance, that is consistent with (and meets criteria for) a bvFTD diagnosis but shows little neuropsychological dysfunction and brain imaging abnormality (Davies et al., 2006; Kipps et al., 2007; Hornberger et al., 2009) and little (if any) progression over a number of years. This causes, in time, uncertainty about the validity of a neurodegenerative explanation of the case. Some investigators believe these individuals have a primary psychiatric diagnosis, such as Asperger syndrome or other developmental syndrome (Hornberger et al., 2009). Yet, despite the imperceptible or slow progression, in some of these patients post-mortem brain examination demonstrates FTLD pathology (Brodtmann et al., 2013). Furthermore, FTD phenocopy patients known to be carriers of the C9orf72 gene mutation associated with FTD (Khan et al., 2012) are considered true variants of FTLD.
Differential diagnosis of FTLD
Several other chapters in this volume review differential diagnosis, including Chapters 2 and 3, which discuss differentiating FTLD from AD, dementia with Lewy bodies (DLB), vascular dementia (VaD), prion diseases, and psychiatric phenocopies. In this chapter we describe general principles in the differential diagnosis of FTLD to attempt to illustrate the process of making an FTLD diagnosis. We also consider a variety of confounders and mimics, some of them rather rare. We aim here to emphasize that clinicians consider a broad swath of conditions when the clinical presentation suggests an FTLD syndrome but is also ambiguous, or when laboratory or neuroimaging investigations include unexpected findings.
Differential diagnosis with diseases traditionally considered “neurologic”
The differential diagnosis of dementia generally includes toxic-metabolic conditions, including anemia, vitamin B12 deficiency, and thyroid, liver, or renal disease. Infectious or inflammatory processes can be a consideration as well. For the most part, these conditions do not produce clinical syndromes overlapping with prototypical bvFTD or PPA, but should always be considered when clinical features are atypical.
Especially in unusually young or unusually rapidly progressive cases, it is worth considering one of the rapidly progressive dementias (RPD), including Creutzfeldt–Jakob disease (which usually progresses to death within 6–12 months) or other prion diseases, viral encephalitis, primary central nervous system vasculitis, paraneoplastic syndromes, or other autoimmune encephalopathies; assessment with a paraneoplastic and vasculitis panel is valuable in these cases.
In young adulthood or even middle age, lipid-storage disorders or white matter degenerative diseases may present with behavioral disturbance and cognitive decline. There are often accompanying features such as retinopathy or organomegaly (e.g., liver or spleen). Mitochondrial disorders may sometimes be included in the differential diagnosis, especially if there are paroxysmal symptoms or accompanying clinical features in the patient and/or maternal family members, including short stature, hearing loss, cardiac conduction loss, diabetes, or retinal disease.
Degenerative basal ganglia disorders can begin with prominent behavioral syndromes. Huntington’s disease often starts as a neuropsychiatric syndrome with obsessive–compulsive and manic symptoms and loss of executive control. Family history should be informative but may be misleading if parental life-span was short or the pedigree is small. Wilson’s disease should be considered in young patients with neuropsychiatric symptoms, in which case a workup for abnormalities of copper metabolism would be appropriate.
Normal pressure hydrocephalus (NPH) is a controversial entity (e.g., McGirr et al., 2013) but should be a consideration, especially in patients with early incontinence, gait disturbance, and a frontal systems behavioral syndrome involving predominantly apathy. In his seminal study of a series of cases of patients with NPH, Raymond Adams (1966) concluded the following: “We watched for [NPH] particularly amongst the patients suspected of having a dementing degenerative disease and found that those in whom poor memory, psychomotor impairment, uncertainty of gait and sphincteric incontinence were early symptoms, and where the evolution of the illness occurred over a few weeks to months and fluctuated, should be suspected as having hydrocephalus rather than presenile or senile brain atrophy.” A prior history of head trauma, subarachnoid hemorrhage, or meningitis increases the suspicion of NPH. Structural neuroimaging should reveal enlarged ventricles in the relative absence of atrophy; in some cases, additional neuroimaging including specialized magnetic resonance imaging (MRI) sequences or fluorodeoxyglucose positron emission tomography (FDG-PET) can also be helpful.
At some hospitals, a three-day lumbar drain trial with repeat cognitive-motor assessments has replaced large-volume cerebrospinal fluid (CSF) collection via lumbar puncture as the assessment for ventriculoperitoneal shunt responsiveness; at the same time, CSF can be examined for other processes including AD-related proteins if AD is suspected. In some cases, shunt treatment can provide meaningful clinical improvement.
The “sagging brain syndrome” is a recently recognized condition thought to involve a chronic CSF leak with downward displacement of brain structures and a cognitive-behavioral syndrome involving frontal systems dysfunction and in some cases symptoms similar to bvFTD. Daytime somnolence and headache are often present. All eight patients in the original report showed evidence of frontotemporal hypometabolism on FDG-PET scans (Wicklund et al., 2011). Identifying the CSF leak is the key to targeted intervention. Some patients improve with treatment including epidural blood patch. Although rare, this condition may be at least partly reversible and so is an important reason to obtain neuroimaging studies in patients with frontal behavioral syndromes.
In our experience, some patients who turn out to be diagnosed with FTD may be thought to have a temporal lobe seizure disorder because of the development of hypergraphia and hyperreligiosity, as well as elevated mood. More commonly, patients with seizure disorders and progressive personality or behavioral changes are suspected of possibly having FTD but in our experience do not exhibit typical symptoms, progressive decline, or typical imaging abnormalities.
Differential diagnosis with diseases traditionally considered “psychiatric”
Patients with FTD are much more likely than those with other neurodegenerative diseases to have received an initial diagnosis of a psychiatric disorder, most commonly depression or bipolar affective disorder, but occasionally schizophrenia (Woolley et al., 2011), OCD, or ADHD. There are, however, important differences in behavior between patients with FTD and those with psychiatric conditions. In FTD, it is unusual for patients to complain of sadness or despair, of anxiety, or indeed to acknowledge any suffering or handicap. Patients with FTD are generally unlikely to notice and regret functional decline, or to show remorse for offensive behavior whereas patients with primary psychiatric states will (with the exception of many with psychosis). In healthcare consultations the spontaneous behavior of FTD patients has been found to differ from that of patients with psychiatric conditions: verbal or physical interruption of the consultation occurred more often in patients with SD and a lack of concern for the clinician’s expectations was more common in FTD patients (Rankin et al., 2008). In our experience, FTLD patients have reached into pockets, attempted to comb the examiner’s hair, sat on the examiner’s table or in the examiner’s chair, and searched for food in the office.
The frequent occurrence of psychotic symptoms in carriers of the C9orf72 mutation merits specific attention. Based on reports from several large series (Dobson-Stone et al., 2012; Kertesz et al., 2013), a substantial minority of C9orf72 mutation carriers present with a psychotic symptom or syndrome, compared with < 4 % of non-mutation carriers (Snowden et al., 2012). These states have included florid mono-delusional psychosis, and bizarre irrational behavior. Not surprisingly, these patients received psychiatric treatment well before referral for neurologic or neuropsychiatric examination. Thus psychosis appearing during or after the fourth decade of life, particularly if characterized by florid mono-delusional beliefs, should arouse suspicion of FTLD and warrants neuropsychiatric consultation. Likewise, psychosis presenting in association with motor phenomena, such as parkinsonism or limb or gait apraxia, should arouse suspicion.
In our experience, many patients eventually determined to have bvFTD, PPA, PSP, or CBS were earlier diagnosed with and treated for depression. Usually apathy and social or occupational withdrawal have been interpreted as a depressive state, and the severity of dysfunction coupled with a dearth of identifiable triggers (social or environmental) prompts a diagnosis of major depression. It should be noted that symptoms seen in major depressive disorder (MDD) such as lack of interest, decreased motivation, anergia, sleep disruption, and impaired concentration (American Psychiatric Association, 2013b) are also observed in FTD. MDD may be distinguished from bvFTD by inquiring about sadness, despair, guilty ruminations, feelings of worthlessness, and suicidal thoughts, symptoms that typically would not appear in FTD (and are thus unlikely to occur together). When sadness is observed in bvFTD or more often PPA, it usually is reactive and linked to an identifiable problem.
Some patients with bvFTD may appear manic and may be diagnosed with bipolar disorder. In particular, the disinhibition and social judgment deficits can overlap with those observed in manic episodes (Woolley et al., 2007). Other common manic symptoms can also be seen in bvFTD, including irritability, distractibility, risk-taking behavior, socially inappropriate behaviors, impaired judgment, and excessive involvement in pleasurable activities with potentially harmful consequences (e.g., compulsive purchases, gambling). Although patients with bvFTD can be inappropriately jocular (Perry and Miller, 2001), patients with euphoric mania have a qualitatively distinct elevated and expansive mood, accompanied by a sense of grandiosity and invulnerability that is uncommon in bvFTD. Individuals with bvFTD also rarely report racing thoughts typically seen in mania, although distractibility could lead to apparent flight of ideas. Most importantly, bvFTD involves an inexorable deterioration of cognitive functions over time, while bipolar disorder is usually an episodic syndrome. Severe cases of bipolar disorder can evolve toward a chronic manic or depressive state, but cognitive impairment is generally not as profound or progressive (Mann-Wrobel et al., 2011).
Further complicating the differential diagnosis of bvFTD versus bipolar disorder, there are a few case reports of autosomal dominant bvFTD preceded by a prodrome of many years of symptoms compatible with primary bipolar disorder. These include dementia secondary to GRN mutations (Cerami et al., 2011) and C9orf72 hexanucleotide repeat expansion (Floris et al., 2013; Meisler et al., 2013). Given that the phenomenologic presentation was identical to bipolar disorder in those cases, a careful family history of FTD or ALS should be performed in patients with late-onset mania, and genetic testing for C9orf72/GRN considered. Conversely, a family history of late-onset mania in a proband with bvFTD might have to be considered as an autosomal dominant mutation. In light of these findings, a genetic link between bipolar disorder and FTD has been suggested, although the rate of C9orf72 mutations in individuals with primary bipolar disorder appears low (Meisler et al., 2013).
OCD may be important to consider in the differential diagnosis, since perseverative, stereotyped, and compulsive/ritualistic behaviors are common in FTD (38–78%), particularly bvFTD and SD (Ames et al., 1994; Mendez et al., 2005). These range from simple stereotypies such as rubbing and tapping, to more complex and elaborate rituals mimicking compulsions typical of OCD (e.g., checking, washing, counting) (Mendez et al., 2005). Cases of late-onset compulsions as the initial manifestation of bvFTD (Mendez et al., 1997; Nakaaki et al. 2007b) and SD (Pompanin et al., 2012) have also been reported.
Given that compulsive behaviors can be similar in bvFTD and OCD, attention should be directed to the qualitative nature of obsessional ideas. Obsessions are recurrent thoughts, urges, or images experienced as intrusive and unwanted, which lead to anxiety and distress. In OCD, the majority of patients engage in repetitive rituals as a response to their obsessions (American Psychiatric Association, 2013b). Behaviors are performed either to reduce distress associated with obsessions, or to prevent an adverse outcome (although there is usually a limited realistic connection between the ritual and the actual outcome). In our experience, patients with FTD and compulsive behaviors typically do not report obsessions – although family members may describe fixations with food, TV programs, cleanliness, object arrangements, and such like that may drive specific compulsions such as food searching, compulsive programming, and so on. Generally, however, FTLD patients usually do not describe or may flatly deny suffering from obsessive thoughts (Ames et al., 1994). It has not been established whether obsessions are indeed absent or whether patients do not have sufficient insight to observe, or lack the capacity to articulate their thoughts in order to report them to clinicians. In those SD patients with the most complex rituals (Modirrousta et al., 2013), a loss of semantic memory might result in an inability to conceptualize or verbalize obsessions.
Hoarding disorder is an OCD-related disorder introduced to the psychiatry lexicon in DSM-5 (American Psychiatric Association, 2013b). It is common in FTD (Mendez and Shapira, 2008), where it may be one of the first symptoms (Nakaaki et al., 2007b). Idiopathic hoarding tends to start in early life, and gets worse over the years. In FTD, hoarding behavior usually follows the onset of cognitive decline, and compared with primary hoarding is associated with less anxiety about the need to save items, and less distress about discarding them. Objects collected by hoarders tend to have at least some theoretical value (e.g., newspapers, clothes, paperwork), and patients usually endorse at least some, often albeit weak, rationale regarding potential use. Items accumulated by patients with FTD can be unsanitary (e.g., rotten food, urine); some authors conceptualize this collecting behavior as secondary to impaired decision-making, without associated rationale of potential future need (Nakaaki et al., 2007a).
We have seen several patients who were ultimately diagnosed with bvFTD after having carried a diagnosis of atypical schizophrenia or a related diagnosis. Overall, schizophrenia and FTD have very different epidemiologic features, particularly average age of onset, and psychotic symptoms such as hallucinations and delusions are rare in FTD (Mendez et al., 2008). However, there is a significant overlap between negative symptoms of schizophrenia and apathy of bvFTD. Negative symptoms are traditionally described with a specific terminology mainly used in the context of psychotic disorders (e.g., avolition, alogia, amotivation), but essentially refer to lack of motivation, initiative, and emotional reactivity, which are core features of apathy (Robert et al., 2009). As such, apathy secondary to FTD and negative symptoms due to schizophrenia may be indistinguishable in themselves. Negative symptoms are common in the prodromal and residual phases of schizophrenia (i.e., when positive symptoms do not dominate), often leading to a progressive withdrawal from social interactions and activities. In addition, there may be overlapping cognitive features. Schizophrenia is associated with mild cognitive deficits in attention, working memory, executive function, declarative memory, and processing speed, which can mimic early deficits of bvFTD (Lewandowski et al., 2011). Disorganized speech and idiosyncratic use of words neologisms could be confused with the lexical and phonologic paraphasias of PPA or some of the clang-association type speech of bvFTD. Interestingly, bvFTD and schizophrenia share common deficits in social cognition (Amodio and Frith, 2006), such as the inability to detect the intent of other persons (theory of mind). Finally, both disorders are usually associated with limited or absent insight.
Given the overlap in negative and cognitive symptoms, the clinical distinction is made based on the presence of positive psychotic symptoms and the long-itudinal history. The classic course of schizophrenia starts with a prodrome between late teens and mid-30s, followed by episodic exacerbations of symptoms, and a variable level of residual deficits. When this longitudinal history with onset prior to 35 is elicited, it is usually sufficient to distinguish it from bvFTD. Differentiating bvFTD from late-onset schizophrenia (i.e., onset after age 40) can be more challenging (Velakoulis et al., 2009).
Social deficits in communication and interpersonal interactions are the core features of autism spectrum disorders (ASD) (American Psychiatric Association, 2013b); given the prominence of social behavioral deficits in bvFTD, it is not surprising that these disorders have been considered to have similar circuit dysfunction. Despite overlap in neuropsychological impairments, differentiating bvFTD from autism spectrum disorder and/or intellectual disability is fairly straightforward since symptoms of the latter two disorders have to be present since early development (American Psychiatric Association, 2013b). When assessing for the first time an adult with impairments suggestive of social cognitive deficits, obtaining a developmental history (including collateral information from family) confirming the appearance of ASD symptoms in childhood and lack of clear progression over time is sufficient to exclude bvFTD. It has been argued that some cases of bvFTD phenocopies (Midorikawa and Kawamura, 2012) could in fact suffer from an undiagnosed mild ASD without accompanying language impairment (previously known as Asperger syndrome) exhibiting increased behavioral disturbance with aging (Hornberger et al., 2008; Midorikawa and Kawamura, 2012). We contend that a diagnosis of ASD should not be made without clear evidence of developmental deficits.
In the early stages of some cases of bvFTD, it can be challenging to ascertain whether distractibility, inattentiveness, and organization deficits could be due to long-standing adult ADHD (diagnosed or undiagnosed) instead of a neurodegenerative process. Although hyperactivity and impulsivity are less common in adult ADHD than inattentiveness (American Psychiatric Association, 2013b), these features also overlap bvFTD (Pose et al., 2013). Restlessness, repetitive movements, and impulsive decision-making can be seen in both conditions. Despite these similarities, there are important phenomenologic distinctions. Repetitive behaviors in bvFTD tend to be stereotyped and perseverative, as opposed to non-specific restlessness and fidgetiness in ADHD. Importantly, adults with ADHD usually have a fair insight about their inattentiveness and/or impulsivity, as opposed to the anosognosia characterizing bvFTD. As with ASD, the key in making this diagnostic distinction is to elicit a history of similar or more severe ADHD symptoms in childhood, starting at least before age 12.
Diagnostic assessment
History of symptoms
It is of vital importance that the history is obtained from a source very familiar with the patient, typically a spouse or other close relative. The history is taken not only with respect to the manifest symptoms, but also in relation to the patient’s lifelong temperament, comportment, and habits. Generally the complaint consists of insidious coarsening of conduct and habits; or it is the gradually progressive loss of speech fluency or comprehension; or it may be worsening self-neglect and abandonment of work, social routines, and relationships (Figure 8.2).
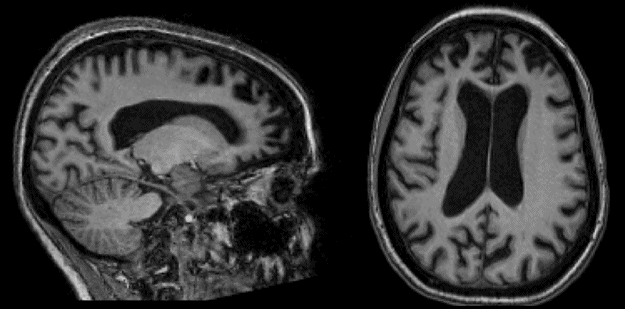
This 49-year-old woman developed gradually progressive difficulty performing the duties of her job and her routine home activities with her family (insidious functional impairment). She had apathy and disinhibition, was impulsive, and no longer seemed to express concern as she had previously done for her daughter’s problems (lack of empathy). There was no obvious memory loss or spatial disorientation. On examination, she had severe impairments in working memory and executive function (with perseveration and difficulties with phonemic verbal fluency). Brain MRI showed prominent prefrontal atrophy on sagittal (left) and axial (right) T1-weighted images. Diagnosis was probable bvFTD.
It is also crucial to interview the source separately, which, while time-consuming, facilitates disclosure and candor, minimizes recrimination from the patient, and provides a “clean” view of the patient’s awareness of the problem. While interviewing the source, the physician might also inquire about that person’s experience of the patient’s illness (i.e., the costs and stresses, and how these are managed), if the source is also a carer or is living with the patient.
By starting the interview with open-ended questioning, the physician will be able to identify the most distressing aspects of the illness, because patients and families will, given the opportunity, usually begin with the most pressing or painful aspects of the problem. From a diagnostic perspective, it is essential to capture the chronology of symptoms, noting the approximate onset of the illness, and the timing, order, and progression of the symptoms. In bvFTD, changes in conduct, habits, activity, and speech typically precede the development of amnesia, disorientation, or apraxia. The illness may also be associated with falls and parkinsonian symptoms, or with muscle wasting and weakness (which suggests coincident ALS and a more malignant prognosis). The physician should also inquire about prior psychiatric history, and probe for symptoms and chronologic patterns (such as lengthy duration, lack of progression, and discrete episodes interspersed with a normal state) that may point to a primary psychiatric disorder rather than FTD. These include chronic paranoia and delusions, prolonged episodes of anxiety and depression, long-standing aloofness and awkwardness (spanning decades rather than a few years), recurring mania, depression, or distressing compulsions (compulsions in FTLD are generally not accompanied by emotional distress).
The possibility of conditions such as hyperthyroidism should also be explored (by inquiring about, for example, heat intolerance, weight loss despite excessive eating, and palpitations) because features such as irritability, restlessness, increased eating (with decreased satiation), and distractibility may mimic FTLD. Finally, careful documentation of disability is the basis for planning immediate and future care. These include handicaps such as disorientation to situations, impaired communication, failures in self-care and grooming routines, abnormal feeding, and loss of bladder and bowel control.
Structure can be imposed on the history-taking process by using specific FTLD symptom inventories (Bozeat et al., 2000; Snowden et al., 2001), the Frontal Behavioral Inventory (Kertesz et al., 1997), or the Neuropsychiatric Inventory (NPI) (Cummings, 1997). Some of these instruments can be given to caregivers in advance as questionnaires, or used to structure an office-based interview. We have developed structured interviews targeting social and language symptoms in FTLD (Sapolsky et al., 2010; Bickart et al., 2014), and instruments have also been developed to quantify disability in FTLD (Knopman et al., 2008; Mioshi et al. 2010; Onyike et al., 2011). Further details on assessment instruments for disability and carer burden and distress are reviewed in Chapter 16.
Related posts:
 Practical management of frontotemporal dementia
The family’s perspective on FTD
Practical management of frontotemporal dementia
The family’s perspective on FTD
 Neuropathology of frontotemporal dementia and related disorders
Neuropathology of frontotemporal dementia and related disorders
 Functional disability and the impact of frontotemporal dementia in everyday life
Historical introduction to FTD
Functional disability and the impact of frontotemporal dementia in everyday life
Historical introduction to FTD
 Overview of frontotemporal dementia and its relationship to other neurodegenerative disorders
Overview of frontotemporal dementia and its relationship to other neurodegenerative disorders
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


