and Aditya G. Shivane1
(1)
Cellular and Anatomical Pathology Level 4, Derriford Hospital, Plymouth, UK
Abstract
This chapter discusses the common congenital malformations and diseases occurring in the perinatal period. Paediatric neuropathology is a highly subspecialised field of neuropathology and a good knowledge of the normal developmental processes and the ability to distinguish normal from abnormal for a particular age is essential for accurate interpretation of gross and microscopic findings. The way the developing nervous system responds to an injury varies during different stages of development. One clinical importance of recognising these conditions (especially malformations) lies in guiding genetic counselling for family members.
Keywords
MalformationsDevelopmentPerinatalHydrocephalusDefects14.1 Congenital Malformations
The CNS malformations contribute to approximately 8–10 % of still births and 5–6 % of early neonatal deaths according to one USA and Europe-wide study [1] and are a significant cause of morbidity. The severity of various malformations form a clinical spectrum with some conditions incompatible with life (e.g. anencephaly) and others causing subtle structural abnormalities resulting in epilepsy, mental retardation, learning and behavioural difficulties (e.g. lissencephaly, microcephaly, cortical dysplasia). Genetic and environmental factors have been found to play a role in the aetiology of CNS malformations.
Before discussing the common congenital malformations, a brief overview of the normal CNS development is presented and outlined in Table 14.1. The nervous system begins to develop with the formation of a neural tube from the midline ectoderm (begins around 16th post ovulation day, and is completed by 4 weeks). This process is termed ‘neurulation’. The neural tube then undergoes segmentation and cleavage to form the major subdivisions of the CNS (which is completed by 8 weeks). The last step is proliferation and migration of cells which eventually populate the various CNS regions (this occurs between 8 weeks and birth; some processes continue even after birth).
Table 14.1
Stages of nervous system development and associated defects
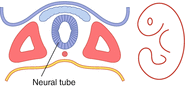 | Neurulation (formation of neural tube; 16th post ovulation day until 4 weeks gestation) | (Neural tube defects) |
Anencephaly | ||
Encephalocoele | ||
Meningomyelocoele | ||
Meningocoele | ||
Spina bifida occulta | ||
 | Segmentation and Cleavage – (formation of major subdivisions of CNS; complete by 8 weeks) | Holoprosencephaly |
Arhinencephaly | ||
Agenesis of corpus callosum | ||
Anomalies of septum pellucidum | ||
 | Proliferation and Migration –(8 weeks- birth or later) | Microcephaly |
Megalencephaly | ||
Lissencephaly | ||
Polymicrogyria | ||
Neuronal heterotopia | ||
Cortical dysplasia |
14.1.1 Neural Tube Defects
Defects of neural tube closure- Craniorachischisis is the most severe form where there is defective closure of much of the neural tube exposing the brain and spinal cord to amniotic fluid, followed by their degeneration and necrosis. Anencephaly results from failure of closure of anterior neuropore. The brain and calvarium are absent and is replaced by a mass of glial and vascularised tissue termed ‘area cerebrovasculosa’. Anencephalics may be alive at birth but invariably die in the new born period. The defect can be detected by ultrasound or MRI scan and is associated with raised alpha-fetoprotein in maternal serum. The incidence is high in Ireland, Wales [2], and India (Punjab), but low in Japan. Folate supplementation in women of child-bearing age has been found to reduce the risk of anencephaly [3]. Other associated abnormalities include hypoplasia of the pituitary, adrenal glands and lungs. Myelomeningocoele shows herniation of spinal cord and meninges through a large vertebral defect and is common at the lumbo-sacral level. Defects above T12 level are more common in females and are associated with other anomalies. Grossly, they can present as flat or cystic mass covered by skin. The histology shows intact or ulcerated, atrophic epidermis with underlying vascularised meningeal tissue containing islands of glial tissue.
Axial defects with herniation of neural tube- Encephalocoele is herniation of brain tissue through a cranial defect, which occurs most frequently in the occipital region. Other less common sites include fronto-ethmoidal and parietal regions. Occipital encephalocoeles may be associated with Meckel-Gruber syndrome; a lethal autosomal recessive condition linked to chromosome 11 and 17, and also has polycystic kidneys, hepatic fibrosis and bile duct proliferation. Protrusion of only the cranial or spinal meninges through a bony defect is termed a Meningocoele. In a spinal meningocoele, both dura and arachnoid herniate; the spinal cord may show hydromyelia, splitting or tethering.
Tail bud defects- Spina bifida occulta is the mildest form of neural tube defect, always a closed defect, which is characterised by minor spinal cord abnormalities such as hydromyelia (over distension of the central canal), diastematomyelia/diplomyelia (longitudinal splitting or duplication of the cord) and tethered cord. These are most common in the lumbo-sacral region.
14.1.2 Chiari Malformations
Chiari malformations are of three types (Table 14.2) and are characterised by cerebellar abnormalities with or without hydrocephalus. The malformation may become symptomatic during infancy, teenage years or in adults. Some of the clinical features include neck or arm pain, sleep-apnoea, stridor, feeding difficulties, nystagmus, lower cranial nerve palsies, weakness and quadriparesis.
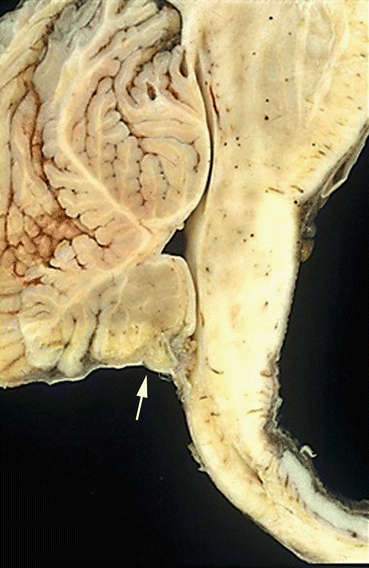
Table 14.2
Chiari malformations
Chiari Type 1 | Chiari Type 2 | Chiari Type 3 |
|---|---|---|
Herniation of cerebellar tonsils through foramen magnum. | Herniation of cerebellar vermis, malformed and downwardly displaced brainstem (Fig. 14.1). | Cerebello-encephalocoele through a bony defect. |
Associated with syringomyelia, skeletal anomalies such as occipital dysplasia and craniosynostosis. | Associated with lumbosacral myelomeningocoele and hydrocephalus. | Associated with brainstem deformities and lumbar spina bifida. |
Fig. 14.1
Chiari type 2 malformation showing herniation of cerebellar vermis (arrow) and elongated brainstem
14.1.3 Disorders of Forebrain Induction
After complete closure of the neural tube, its anterior end undergoes segmentation into three vesicles- prosencephalon (which gives rise to forebrain), mesencephalon (midbrain) and rhombencephalon (hindbrain). The forebrain and hindbrain undergo further divisions into telencephalon (cerebrum), diencephalon (basal ganglia and thalamus) and metencephalon (pons and cerebellum), myelencephalon (medulla) respectively. Subsequent cleavage of the prosencephalon gives rise to two cerebral hemispheres and ventricles. Several genes such as Shh (sonic hedgehog), Bmp7 (bone morphogenetic protein 7) and Emx1 play important roles in forebrain development [4–6].
Holoprosencephaly (single ventricle and single cerebrum) results from failure or incomplete cleavage of the prosencephalon. This can be sporadic or associated with trisomy’s 13, 18 and triploidy. Severely affected children often die in the neonatal period. In those who survive, clinical symptoms include mental retardation, seizures, anosmia and pituitary insufficiency. Based on the severity of the malformation the following different forms are recognised: Arhinencephaly (mildest), lobar, semi lobar and alobar holoprosencephaly (severe form). In arhinencephaly, the olfactory bulbs, tracts and gyri recti are absent on both sides. In lobar form, the cerebral hemispheres are separated by inter hemispheric groove, but the cingulate cortex is continuous along the midline. The corpus callosum and olfactory bulbs are still absent or hypoplastic. The semi lobar form (Fig. 14.2) shows partially formed shallow inter hemispheric fissure, mainly in the posterior occipital regions. Anteriorly, there is continuity of the cingulate cortex across the midline and olfactory bulbs are usually absent. The alobar form is severest form associated with various facial anomalies. This is characterised by a globular undivided cerebrum with monoventricle, fused basal ganglia and thalami, and no olfactory structures or corpus callosum. Histology may reveal abnormalities in cortical cytoarchitecture such as polymicrogyria and neuronal heterotopias.
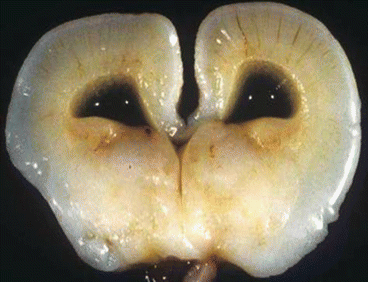
Fig. 14.2
Semi lobar holoprosencephaly showing absence of corpus callosum, dilated lateral ventricle, fused thalami and absent third ventricle
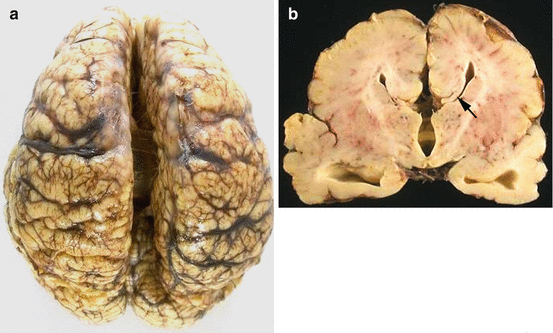
Agenesis of corpus callosum can be total or partial (posterior portion, splenium, absent). It can be sporadic or familial and associated with other malformations such as holoprosencephaly. The cingulate gyrus is also deficient. The corpus callosum is replaced by an abnormal longitudinal bundle (called the ‘Probst bundle’) and the angles of the lateral ventricles are upturned (‘bat wing’ appearance) (Fig. 14.3a, b).