Syndrome
Relevant antibodies
CNS
Subacute cerebellar degeneration 25 %
Hu, Yo, CV2/CRMP5, Ri, Tra, amphiphysin, VGCC
Encephalomyelitis 6 %
Hu, CV2/CRMP5, amphiphysin
Limbic encephalitis 10 %
Hu, Ma2, CV2/CRMP5, Ri, amphiphysin
NMDAR, Lgi1b, CASPR2b, GABA(b)-R, AMPA-R, mGluR5, glyRb, GADb
Opsoclonus-myoclonus syndrome (adults) 2 %
Ri, Hu, Ma/Ta, NMDAR
Retinopathy 1 %
Hu, CV2/CRMP5, recoverin
Stiff-person syndrome 1 %
Amphiphysin, glyRb, GADb
Table 21.2
Onconeural antibodies found in paraneoplastic syndromes
Antibody | Antigen | Associated syndromes and symptoms | Most common tumours |
|---|---|---|---|
Onconeural antibodies (well-characterised, paraneoplastic antibodies tumour in >90 %) | |||
Anti-Hu (ANNA-1) | HuD | Encephalomyelitis, limbic encephalitis, cerebellar degeneration, brainstem encephalitis, multi-segmental myelitis, sensory neuronopathy, sensory-motor neuropathy, autonomic neuropathy | Lung cancer (85 %), mostly SCLC, neuroblastoma, prostate carcinoma |
Anti-Yo (PCA-1) | CDR2, CDR62 | Paraneoplastic cerebellar degeneration | Ovarian, breast cancer |
Anti-CV2/CRMP5 | CRMP5 | Encephalomyelitis, polyneuropathy, optic neuritis, limbic encephalitis, choreatic syndromes, cerebellar degeneration | SCLC, thymoma |
Anti-Ta/Ma2a | MA-proteins | Limbic encephalitis, rhombencephalitis, m>>f | Testicular cancer |
Anti-Ri (ANNA-2) | NOVA-1 | Opsoclonus-myoclonus syndrome, rhombencephalitis, cerebellar degeneration, myelitis, jaw dystonia, laryngospasm | Breast, ovarian carcinoma, SCLC |
Anti-amphiphysin | AMPHIPHYSIN | Stiff-person syndrome, limbic encephalitis, rhombencephalitis, cerebellar degeneration, polyneuropathy | Breast cancer, SCLC |
Anti-recoverin | RECOVERIN | Retinopathy | SCLC |
Anti-SOX-1 (AGNA) | SOX-1 | Non syndrome-specific | Sensitivity 67 %, specificity 95 % for SCLC in LEMS |
Partially characterised onconeural antibodies (antigen not characterised or positive predictive value for tumour unknown) | |||
Anti-Tr (PCA-Tr) | DNER | Cerebellar degeneration | Hodgkin lymphoma, non-Hodgkin lymphoma |
Anti-Zic4 | ZIC1-4 | Cerebellar degeneration | SCLC |
PCA-2 | 280 kD | Encephalitis, Lambert-Eaton myasthenic syndrome, polyneuropathy | SCLC |
ANNA-3 | 170 kD | Neuropathy, cerebellar degeneration, limbic encephalitis | SCLC |
The probability of a PNS depends on (1) the occurrence of a classical or nonclassical syndrome, (2) absence or presence of a detectable tumour and (3) detection of a well-characterised antibody, partially characterised antibody or absence of known antibodies (Fig. 21.1). Classical syndromes and detectable tumours or any syndrome and detectable well-characterised antibodies can be classified as definite PNS. A nonclassical syndrome associated with a malignancy is considered to represent a definite PNS if either a well-characterised or partially characterised antibody can be detected or the neurological sign and symptoms respond to treatment of the underlying malignancy. Other situations can be classified as possible PNS after exclusion of differential diagnoses.
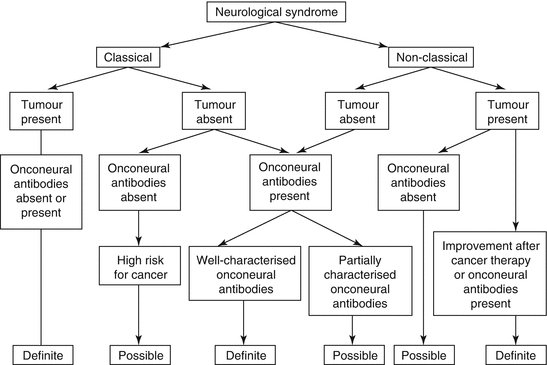
Fig. 21.1
Flow chart showing the level of diagnostic evidence of a paraneoplastic neurological syndrome depending on the neurological syndrome, the presence of absence of onconeural antibodies and a tumour. Of note, a definite and possible PNS without detection of a tumour requires regular tumour screenings for 4 years (Reprinted with permission from Graus et al. (2004))
In most patients with PNS, neurological symptoms precede tumour diagnosis, because tumours are too small to be detectable with available techniques. If any classical or nonclassical syndrome co-occurs with a well-characterised onconeural (e.g. Hu, Yo) antibody, initial tumour screening should be carried out according to recently published European guidelines and close oncological follow-up every 3–6 months for 4 years is warranted (Titulaer et al. 2011a). A similar tumour screening approach is advisable for classical syndromes without well-characterised or with partially characterised antibodies. If the syndrome is nonclassical and no antibodies are found, the method of tumour screening and surveillance depends on the level of clinical suspicion and no clear guidelines exist. It is good clinical practice to perform close clinical follow-up visits and possibly repeat tumour screening but alternative diagnoses have to be considered frequently.
21.3 Pathophysiology
In most paraneoplastic neurological syndromes described in this chapter, ectopic expression of neuronal antigens by systemic tumours drives an immune-mediated inflammatory response against central or peripheral nervous tissue. These onconeural antigens can be expressed by many tumours and most often by tumours of neuroectodermal lineage, e.g. small-cell lung cancer (SCLC). Antitumour immune responses are a common phenomenon, e.g. low titre anti-Hu antibodies are detectable in approximately 16 % of patients with SCLC without neurological symptoms (Dalmau et al. 1990; Graus et al. 1997). However, only a minority of these develop a paraneoplastic neurological syndrome. This might be due to intrinsic tumour factors (degree of inflammation, downregulation of HLA) (Maverakis et al. 2012), which causes breach of tolerance to self antigens. Furthermore, host factors like human leucocyte antigens (HLA) haplotypes likely contribute (Dalmau et al. 1995), but the main factors remain unclear.
Recent years have shown that the subcellular localisation of the detected antigen plays a major role for disease mechanisms. As already mentioned, onconeural antibodies are directed against intracellular antigens. These are not directly accessible to the antibodies. Most likely, the main pathogenic effect is carried out by cytotoxic T cells, resulting in neuronal cell death (Lancaster and Dalmau 2012). There are some exceptions to this rule. Amphiphysin antibodies might have a direct pathophysiological effect as suggested by in vivo models although the antigen is localised intracellularly (Sommer et al. 2005). Conversely, antibodies directed against neuronal surface proteins, δ/notch-like epidermal growth factor-related receptor (DNER), also known as anti-Tr (Greene et al. 2014; de Graaff et al. 2012), and antibodies against metabotropic glutamate receptor 5 (mGluR5) (Lancaster et al. 2011; Graus et al. 2014) can be found in PCD and LE in association with Hodgkin lymphomas (HL), respectively. However, beyond their atypical cellular localisation, the pathophysiology of anti-Tr and anti-mGluR5 antibody-associated PNS might differ with respect to cause of the autoimmune response as, in contrast to other onconeural antibodies, these antigens are not expressed in the underlying tumours (Graus et al. 2014). Nevertheless, this distinction helps to differentiate the paraneoplastic syndromes described in this chapter from the therapy-responsive encephalitis syndromes associated with neuronal surface and synaptic antibodies described in Chap. 6.
In Lambert-Eaton myasthenic syndrome (LEMS), antibodies directed against presynaptic P/Q-type voltage-gated calcium channels (VGCC) directly interfere with proper depolarisation-dependent acetylcholine release within the neuromuscular junction (Titulaer et al. 2011b). In paraneoplastic LEMS, the underlying SCLCs express VGCC (Roberts et al. 1985). However, as antibodies against VGCC also occur in non-paraneoplastic LEMS, which present about 50 % of the cases (Titulaer et al. 2011b), VGCC antibodies are not regarded as onconeural antibodies.
Not all paraneoplastic syndromes involving the nervous system are induced by an immune attack against structures of the nervous system. A notable exception is the POEMS syndrome. This acronym stands for polyneuropathy, organomegaly, edema, M-gradient and skin changes (Li and Zhou 2013). This subtype of monoclonal gammopathy-associated neuropathy normally shows an M-gradient representing IgA lambda. The assumed pathophysiology is a massively increased vascular endothelial growth factor (VEGF) contents in the patient’s platelets somehow induced by the neoplastic plasma cells. VEGF is thought to induce a breakdown of the blood-nerve barrier with subsequent influx of serum proteins and demyelination. Highly increased serum VEGF levels are diagnostic.
21.4 Classical and Nonclassical Paraneoplastic Neurological Syndromes
21.4.1 Progressive Cerebellar Degeneration
PCD is one of the paraneoplastic syndromes with onconeural antibodies most frequently encountered in clinical practice (Giometto et al. 2010). Typical PCD clinically presents as a pancerebellar syndrome with both truncal and appendicular ataxia. Although initially, there can be asymmetrical involvement of the limbs, during the course of the disease the ataxia becomes symmetrical. Disability is usually severe so that most patients become unable to walk or even sit without support. Most patients have dysarthria, in part to an extend that speech becomes unintelligible, and nystagmus (Anderson et al. 1988a). Neurological symptoms progress over a few weeks or months and eventually stabilise, unfortunately most often with severe residual and permanent symptoms due to cerebellar dysfunction (Anderson et al. 1988a). Most patients have other signs of neuronal involvement beyond the cerebellum including extensor plantar responses, hyporeflexia or mild cognitive dysfunction. In some cases, progressive involvement of neuronal structures other than the cerebellum is observed leading to the diagnosis of progressive encephalomyelitis.
In an initial series, approximately 50 % of patients with PCD had detectable antineuronal antibodies upon screening using immunohistochemistry (Anderson et al. 1988a). In a later series of patients positive for antineuronal antibodies, five different antibody specificities were identified in PCD (Shams’ili et al. 2003): anti-Yo antibodies were most frequent, closely followed by anti-Hu antibodies. Anti-Tr (recently identified as directed against DNER) and anti-Ri occurred less often and anti-mGluR1 antibodies were rare. Another well-defined onconeural antibody detected in some cases of PCD is anti-CV2/CRMP5 (Honnorat et al. 1996). In addition, serum and CSF of patients with PCD and SCLC often harbours antibodies against VGCC (Graus et al. 2002) and in some cases against Zic4 (Bataller et al. 2004). Patients with PCD and VGCC antibodies should be examined for the presence of Lambert-Eaton myasthenic syndrome (Mason et al. 1997). One of four cases of anti-Ca/anti-ARHGAP26 antibodies and PCD has been reported to be paraneoplastic because of the coincidence with ovarian cancer (Jarius et al. 2013). In addition, two patients with antibodies against protein kinase Cγ, which is highly expressed in Purkinje cells, and cancer have been reported (Sabater et al. 2006; Hoftberger et al. 2013).
The severity of clinical impairment due to cerebellar dysfunction correlates with the antibodies detected. Compared to patients with anti-Yo, anti-Hu and anti-Tr, patients with anti-Ri antibodies retain their ability to walk far more often (Shams’ili et al. 2003). Anti-Ri antibodies were also associated with a lower incidence of dysarthria and nystagmus while anti-Hu positivity was associated with an increased incidence of additional neurological signs and symptoms. The median survival of patients with anti-Yo and anti- Hu antibodies and PCD was 13 and 7 months, respectively. This was much shorter than the survival in the group of patients with anti-Tr antibodies (>113 months). Also patients with anti-Ri-associated PCD tended to live longer. Interestingly, in a subset of patients with anti-Ri and anti-Tr antibodies, symptoms can improve considerably upon treatment (Shams’ili et al. 2003; Bernal et al. 2003), which only rarely occurs in anti-Yo-positive patients (Shams’ili et al. 2003).
Upon histopathological examination, PCD is characterised by a severe and diffuse loss of Purkinje cells throughout the cerebellum associated with CD8+ T-cell infiltrates and microglial activation throughout the brain (Giometto et al. 1997; Storstein et al. 2006).
Usually the neurological syndrome stabilises after several months but functionally on a very poor level with 90 % of patients being wheelchair bound (McKeon et al. 2011a). On magnetic resonance imaging (MRI) early during the disease, usually no abnormalities are found. Within months, atrophy may develop (McKeon et al. 2011a). In a few cases, fluor-deoxyglucose-positron-emission-tomography (FDG-PET) showed cerebellar hypermetabolism early during the disease (Choi et al. 2006).
The differential diagnosis of PCD is broad. Cerebellar ataxia can evolve as a predominant symptom in many neurological diseases, e.g. spinocerebellar ataxias, multiple system atrophy and non-paraneoplastic immune-mediated cerebellitis associated with antibodies against glutamic acid decarboxylase (GAD). However, paraneoplastic cerebellar degeneration is distinguished from these disorders by its relentless, subacute progression. In addition to its slower progression and indolent course, anti-GAD-positive cerebellar degeneration is also often associated with other autoimmune diseases including diabetes mellitus, thyroiditis and sometimes vitiligo (Honnorat et al. 2001). In some cases, Creutzfeldt-Jakob disease (CJD) can mimic PCD (Grau-Rivera et al. 2014). Cerebellitis of infectious origin, e.g. varicella zoster virus, can initially mimic PCD; however, it usually occurs in children (Bozzola et al. 2014). In rare occasions, HIV infection can induce progressive cerebellar ataxia (Tagliati et al. 1998) or it is a caused by infratentorial progressive multifocal leucoencephalopathy in the context of AIDS (Ali et al. 2013) or immunosuppression (Jones et al. 1982). The initial phase of Wernicke-Korsakoff syndrome due to thiamine deficiency can be dominated by subacute ataxia (Butterworth 1993).
21.4.2 Subacute Sensory Neuronopathy
Subacute sensory neuronopathy (SSN) is by far the most frequently encountered PNS affecting the peripheral nervous system (Giometto et al. 2010). This unique syndrome was initially described by Denny-Brown in two cases with neuropathy associated in bronchial carcinoma in 1948 (Denny-Brown 1948). Patients with SSN are characterised by subacutely spreading, frequently asymmetrical, non-length-dependent numbness and paraesthesias, including the face and trunk, which are associated with severe sensory ataxia. About half of the patients report neuropathic pains. Clinically, there is no motor involvement. Due to sensory ataxia, most patients become unable to walk without aid. In addition, frequently dysautonomia is observed. Just recently diagnostic criteria were published, which rely upon the characteristic clinical picture as well as on the typical result obtained in nerve conduction studies (Camdessanche et al. 2009). These reflect the pathophysiology with loss or reduced amplitude of sensory compound nerve action potentials. However, clinically inapparent involvement of motor axons with decreased amplitudes and nerve conductions velocity especially in the lower limb can be observed (Camdessanche et al. 2009). Sensory-evoked potentials are lost in most cases and abnormal when detectable (Camdessanche et al. 2009). Interestingly, upon electrophysiological examination, the masseter reflex in patients with SSN and facial hypaesthesia is unaffected while the blink reflex is abnormal (Valls-Sole et al. 1990). This observation was explained by the fact that perikaryons carrying the input from the masseter spindles are not localised in the trigeminal ganglion, which is affected by the disease. In contrast, they reside within mesencephalic nucleus of the trigeminal nerve. Thus, they are part of the central nervous system (Ongerboer de Visser 1983).
Approximately 80 % of patients with paraneoplastic SSN harbour anti-Hu antibodies. Other onconeural antibodies usually do not associate with SSN (Molinuevo et al. 1998). Small-cell carcinomas of the lung (SCLC) are by far the most common tumour found in anti-Hu-positive SSN. These are also frequently found in paraneoplastic SSN without known antibodies. In addition, a wide variety of cancers may be associated with SSN, including prostate, breast, pancreatic, neuroendocrine, bladder and ovarian cancer (Rudnicki and Dalmau 2005).
At autopsy, histological examination in SSN shows inflammatory infiltrates including CD8+ T cells in the spinal and autonomic ganglia (Panegyres et al. 1993; Wanschitz et al. 1997).
SSN is distinguished clinically from other forms of predominantly sensory neuropathy by the non-length-dependent development of sensory symptoms. Thus, sensory disturbances frequently involve the upper limb and may also include the face. In addition, sensory involvement is more often asymmetrical and presents predominantly as sensory ataxia. Finally, it is characterised by pronounced axonal loss upon sensory nerve conduction studies in the upper limbs while only minor changes can be found in motor nerve conduction studies (Camdessanche et al. 2009). Non-paraneoplastic SSN can evolve in the context of systemic autoimmune diseases, most often Sjögren’s syndrome (Valls-Sole et al. 1990; Griffin et al. 1990). In addition, toxic SSN can occur upon chemotherapy including cisplatin (Gill and Windebank 1998) administered to cancer patients (Krarup-Hansen et al. 2007).
21.4.3 Limbic Encephalitis
The association of LE and cancer was initially described in 1968 by Corsellis et al. (1968). Diagnostic criteria, although not formally validated, include the following criteria: (a) a clinical picture with short-term memory loss, psychiatric symptoms or epileptic seizures suggesting the involvement of limbic structures; (b) an interval <4 years between onset of symptom and the diagnosis of cancer; (c) exclusion of other cancer-related explanations including metastasis, infections, nutritional or metabolic disturbances, cerebrovascular disease or side effect of cancer therapy; and (d) finally, either inflammatory cerebrospinal fluid (CSF) changes, unilateral or bilateral temporal hyperintensities in T2-/FLAIR-weighted MRI or temporal atrophy on T1-weighted images or EEG showing slow or sharp-wave activity in one or both temporal lobes need to underscore either the inflammatory origin or involvement of limbic structures (Gultekin et al. 2000).
In a recent European series, LE occurred in approximately 10 % of all PNS (Giometto et al. 2010). The neurological symptoms evolved in half of the patient before the diagnosis of cancer (Gultekin et al. 2000). Most cases present with short-term memory loss (84 %), seizures (50 %), acute confusional states (46 %) and/or and psychiatric symptoms (42 %) with affective symptoms, hallucinations and personality changes being most common. Symptoms usually evolve within days and weeks rather than months and years. A subset of patients also develops signs of hypothalamic dysfunction including hyperthermia, weight gain, endocrine dysfunction including diabetes insipidus and hypersomnia. In a few patients, cerebellar or brainstem involvement or neuropathy can occur (Gultekin et al. 2000). Especially in patients with anti-Hu antibodies, the syndrome can eventually evolve into an encephalomyelitis (Graus et al. 2001) (see below).
Anti-Hu antibodies most often associate with SCLC and the prognosis is generally unfavourable. In patients with anti-CV2/CRMP5 antibodies, optic neuritis and chorea can be associated features (Honnorat et al. 1996; Cross et al. 2003). Other onconeural antibodies are anti-Ma2 (also known as anti-Ta) and anti-amphiphysin. Most commonly associated tumours are lung cancers, especially SCLC. However, in women, breast and ovarian cancer are common, and in young male patients, one has to consider testicular cancer (anti-Ma2) (Voltz et al. 1999). Rarely, neuroectodermal skin cancer (Merkel-cell carcinoma) has been described (Greenlee et al. 2002). Furthermore, thymomas and lymphomas have to be considered in onconeural and neuronal cell-surface antibody-associated syndromes (Yu et al. 2001; Lancaster et al. 2011; Ingenito et al. 1990; Irani et al. 2012) (mGluR5, Chap. 6). Importantly, limbic encephalitis can be associated with onconeural (e.g. Hu) and neuronal cell-surface antibodies (e.g. against AMPA receptors, GABAB receptors, Chap. 6) or a combination of antibodies from both groups (e.g. Hu and GABAB receptors) (Lancaster and Dalmau 2012). Of the cases with paraneoplastic limbic encephalitis previously considered as seronegative, up to 40 % might harbour anti-GABAB receptor antibodies (Boronat et al. 2011) (Chap. 6). In summary, in LE it is important to comprehensively test CSF and serum for known and unknown onconeural and neuronal surface antibodies. In cases with definite LE with well-characterised antibodies, about 50 % of the patients died during follow-up, and the minority showed improvement in response to therapy (Bataller et al. 2007). Patients with LE and neuronal surface or synaptic autoantibodies have a far better prognosis (Chap. 6).
In 70–80 % of cases with paraneoplastic LE, neuroimaging studies show medial temporal lobe hyperintensity on fluid-attenuated inversion recovery and T2-weighted images (Gultekin et al. 2000; Lawn et al. 2003). Upon neuropathological examination, brains of patients with classical paraneoplastic LE show neuronal loss, inflammatory infiltrates and microglial nodules in limbic structures including the hippocampus and amygdala (Alamowitch et al. 1997; Newman et al. 1990).
In oligosymptomatic cases of LE, schizophrenia or other psychiatric diseases might be considered as differential diagnosis. In addition, encephalopathy due to intoxication or metabolic disturbance might mimic some aspects of LE as does temporal lobe epilepsy or status epilepticus of complex-partial seizure due to other pathophysiologies. Infectious aetiologies, most importantly herpes simplex encephalitis have to be excluded (Granerod et al. 2010). Human herpes virus type 6 (HHV6) can induce LE-like encephalitis, including typical neuroimaging results, in patients after allogenic haematopoietic stem cell transplantation (Bhanushali et al. 2013). LE with antibodies against neuronal surface or synaptic proteins (Chap. 6) are more common than the paraneoplastic LE variants described here (Granerod et al. 2010).
21.4.4 Encephalomyelitis
PCD, SSN as well as paraneoplastic LE can show signs and symptoms suggesting involvement of other parts of the nervous system. Thus, there is a spectrum from more localised syndromes (PCD, LE, SSN) to a widespread affection of the entire nervous system coined encephalomyelitis (EM) (Henson et al. 1965). The latter is characterised by a simultaneous or progressive involvement of different subsystems of the nervous system including the peripheral nervous system. This spectrum is best described in anti-Hu-associated syndromes with underlying SCLCs. Here, one quarter of patients show isolated involvement of the nervous system, while in three quarters of patients either two or three different areas of the nervous system are affected (Dalmau et al. 1992). SSN predominates in about 60 % of the cases, but motor neuron dysfunction, limbic involvement, cerebellar degeneration, brainstem encephalitis and autonomic dysfunction can also be dominating clinical features (10–20 %) (Dalmau et al. 1992). The distinction between encephalomyelitis and localised syndromes, e.g. PCD with minor brainstem involvement, remains somewhat arbitrary. According to the recommended diagnostic criteria, the diagnosis of EM should be avoided in cases with a clearly dominant localised syndrome (Graus et al. 2004) (Fig. 21.2). Nevertheless, the simultaneous or consecutive association of subacute dysfunction of different areas of the nervous system is highly suggestive of a paraneoplastic origin, and thus its recognition as (EM) is helpful for clinical practice.
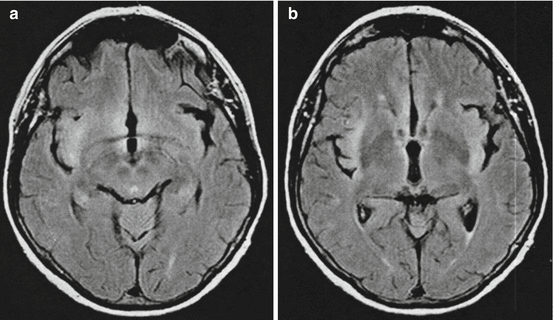
Fig. 21.2
Subclinical involvement of limbic structures in a patient with anti-Hu-associated subacute sensory neuropathy. The 69-year-old female with subacute sensory neuropathy (SSN) and high-titre anti-Hu antibodies presented with the typical clinical picture of SSN with paraesthesias of the limbs and severe sensory ataxia. In addition, mild neuropsychiatric abnormalities including apathy and mental slowness were noted. Cranial MRI (FLAIR) showed hyperintensity of the temporal lobe (a), insular region (a/b) and the gyrus cinguli (b). Although these findings proved that the paraneoplastic syndrome affected areas of the nervous system beyond the spinal ganglia, the diagnosis of SSN not encephalomyelitis was made as the SSN was by far the predominant paraneoplastic syndrome. Although asbestosis was present, no lung tumour could be detected. After short-lived stabilisation in response to high-dose steroids, the patient’s conditions deteriorated and she died 5 months after onset of symptoms
Most patients suffering from (EM) have anti-Hu antibodies (Graus et al. 2001; Dalmau et al. 1992). However, (EM) was also reported in single cases with anti-CRMP5 antibodies (Honnorat et al. 1996). Although sharing parts of the name, the syndrome of progressive encephalomyelitis with rigidity and myoclonus, which is associated with a different subset of antibodies, is clinically more related with stiff-person syndrome and thus is discussed below.
21.4.5 Opsoclonus-Myoclonus Syndrome
The paraneoplastic opsoclonus-myoclonus syndrome (OMS), also named Kinsbourne syndrome after its first description in 1962 (Kinsbourne 1962), has a characteristic age distribution. Each age group has specific associated antibodies, underlying tumours and prognosis. In paediatric cases, the mean onset is 18–20 months with only 13 % of cases being older than 2 years (Boltshauser et al. 1979; Talon and Stoll 1985). Most of these patients suffer from neuroblastomas but onconeural antibodies are extremely rare. Young adults (median 22 years) have either idiopathic or teratoma-associated OMS and novel neuronal surface antibodies can be found in some of them (Armangue et al. 2014). Adult paraneoplastic cases associated with other tumours were older than 40 years with a mean age of onset of 66 years reported (Anderson et al. 1988b; Bataller et al. 2001). Generally, paraneoplastic OMS is rare and occurs approximately tenfold less often than PCD or SSN (Giometto et al. 2010).
Opsoclonus is an ocular movement disorder defined by continuous, irregular and conjugated chaotic back-to-back saccades in all direction but preferentially horizontally without intersaccadic interval. Opsoclonus is normally increased by eye closure and fixation and persists during sleep (Bellur 1975). In OMS, opsoclonus is typically associated with action myoclonus of the extremities; also, asterixis may occur (Caviness et al. 1995). The neuronal generators of opsoclonus and myoclonus are distinct as there is no temporal association. In addition, opsoclonus and myoclonus are often associated with ataxia, which in comparison to PCD has been reported to be more truncal than appendicular (Anderson et al. 1988b). However, in many cases the presence of ataxia is difficult to ascertain due to severe myoclonus. Thus, its presence has even been questioned with the ataxic movement disorder interpreted rather as a result of the myoclonus (Pranzatelli 1992). The onset of OMS is usually subacute within days or weeks. Initially, patients frequently complain of vertigo. Both oscillopsias due to opsoclonus and stance and gait instability due to myoclonus (and ataxia) can become so severe that the patients lose their ability to walk unaided and prefer to lie supine with their eyes closed (Pranzatelli 1992). Encephalopathy has been reported repeatedly in OMS with altered mental status in the sense to apathy, lethargy or confusion occurring in approximately half of adult cases and progressing to stupor or coma in a minority of these. Dysphagia and dysarthria may be present (Pranzatelli 1992). Children that survive OMS frequently have neurological, neurocognitive and behavioural deficits, which can be severe in some cases (Hayward et al. 2001). If the tumour is not treated, patients with adult paraneoplastic OMS usually die from encephalopathy; antineoplastic treatment can improve symptoms considerably (Bataller et al. 2001).
Only a minority of both adult and paediatric patients with paraneoplastic OMS harbour antineuronal antibodies (Bataller et al. 2001; Antunes et al. 2000). The most common antibody found to be associated with adults paraneoplastic OMS is anti-Ri, which in a review of published cases occurred in 66 %. Much less often, anti-Hu and anti-amphiphysin as well as VGCC antibodies are found (Klaas et al. 2012). Neuronal surface antibodies of unknown specificity have been observed in young adult patients with teratomas or testis tumours (Armangue et al. 2014). In single cases, OMS might be part of the clinical presentation of NMDA receptor-antibody-associated encephalitis (Kurian et al. 2010; Smith et al. 2011); however, no tumour has been detected in these two cases, a patient with OMS and GABAB receptor antibodies (DeFelipe-Mimbrera et al. 2014) and OMS with GAD antibodies (Bhandari 2012). In addition, diverse autoantigens, including proteins of the postsynaptic density, have been found in paraneoplastic and non-paraneoplastic OMS but currently do not have diagnostic relevance (Bataller et al. 2003). In paediatric paraneoplastic OMS, cases with onconeural antibodies showed anti-Hu (Antunes et al. 2000; Fisher et al. 1994). In addition, antibodies against neurofilament (Connolly et al. 1997) and the cell surface of granule cells (Blaes et al. 2005) have been reported in paediatric OMS. However, the clinical or pathophysiological relevance of these findings remains to be explored.
Diverse tumours have been found associated with adult paraneoplastic OMS (Pranzatelli 1992). Lung cancer is the most frequent cancer followed by breast cancer (Klaas et al. 2012). Paediatric cases are associated with neuroblastoma, ganglioneuroblastoma or ganglioneuroma (Pranzatelli 1992). About 2 % of children with neuroblastomas develop OMS, and this is associated with a more favourable prognosis of the tumour (Altman and Baehner 1976).
Neuroimaging studies most frequently reveal pontine lesion in adult OMS (Kim et al. 2009; Hattori et al. 1988). Children with paraneoplastic OMS usually develop persisting cerebellar atrophy (Hayward et al. 2001) (Fig. 21.3).
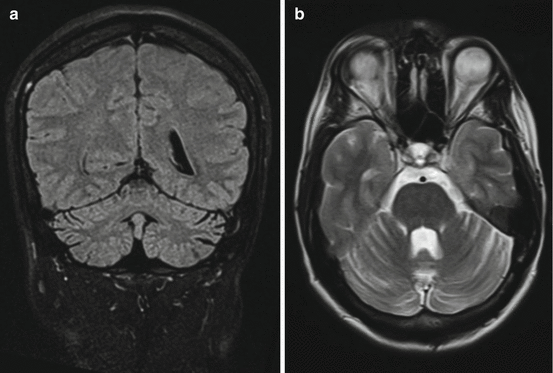
Fig. 21.3
Cerebellar atrophy as a residuum of childhood opsoclonus-myoclonus syndrome associated with neuroblastoma. Cranial MRI: (a) coronar FLAIR and (b) horizontal T2-weighted images. The 30-year-old female developed opsoclonus-myoclonus syndrome (OMS) at the age of 18 months. Several months later, a neuroblastoma of the left adrenal gland was diagnosed and surgically removed. After surgery and steroid therapy, the patient rapidly improved. However, cerebellar ataxia and behavioural-developmental deficits remained. Although the patient became ambulatory, frequent falls persistent. The patient is independent regarding activities of daily living; however, no school-leaving graduation could be obtained
Autopsy findings in paraneoplastic OMS did not reveal any specific abnormalities. Mostly, they report diffuse microglial proliferation and lymphocytic infiltrates in different brain areas including the cerebellum and brainstem (Anderson et al. 1988b; Young et al. 1993). However, mild Purkinje cell loss has been reported in some cases (Anderson et al. 1988b).
In adults, only a minority of cases with OMS are paraneoplastic (Klaas et al. 2012), whereas up to 40 % of children with OMS have neuroblastomas or related tumours (Brunklaus et al. 2012). The rest remains idiopathic or infectious, parainfectious or toxic/metabolic (Pranzatelli 1992; Klaas et al. 2012). Infectious causes include HIV seroconversion, poliomyelitis, EBV, CMV, mumps, HHV6, West Nile virus and others (Pranzatelli 1992; Klaas et al. 2012; Belcastro et al. 2014; BirluTiu and BirluTiu 2014). A number of intoxications, including amitriptyline, lithium and carbamazepine, were found to induce OMS (for a more complete list, compare (Pranzatelli 1992)). The clinical course of non-paraneoplastic OMS is usually monophasic and, if the underlying infection is not life threatening, mostly benign.
21.4.6 Chronic Intestinal Pseudo-obstruction
Chronic intestinal pseudo-obstruction (CIPO) is subsumed under dysautonomia in epidemiological overviews about PNS (Giometto et al. 2010). Dysautonomia is a rather rare feature in PNS occurring approximately fivefold less often than SSN and as often as LEMS (Giometto et al. 2010). Thus, it can be concluded that paraneoplastic CIPO is even rarer. The first study indicating that CIPO can occur in the context of a PNS was reported in 1978 (Ahmed and Carpenter 1975). Soon, this was followed by larger case series of patients with SCLC or, in rare occasions, pulmonary carcinoids (Chinn and Schuffler 1988).
Per definition, CIPO is a clinical syndrome presenting with symptoms and signs of intestinal obstruction in the absence of mechanical blockade (Schuffler et al. 1981). Frequent complaints of patients with paraneoplastic CIPO are nausea and vomiting, abdominal pain and distension and severe obstipation. In some patients, additional neurological symptoms occur including other signs of dysautonomia including neurogenic bladder and orthostatic hypotension. Also peripheral neuropathy, cognitive disturbances or ataxia have been noted (Chinn and Schuffler 1988).
In a large series of 162 patients with PNS and anti-Hu antibodies, approximately 10 % presented with solely gastrointestinal symptoms (Lucchinetti et al. 1998). Some patients with medium to high titres of antibodies against α3 acetylcholine receptor antibodies develop limited dysautonomia with gastrointestinal dysmotility (McKeon et al. 2009).
The most frequent tumour associated with CIPO is SCLC (Lucchinetti et al. 1998; Lennon et al. 1991). CIPO has also been reported in a younger patients in association with neural crest tumours (Gohil et al. 2001), e.g. a ganglioneuroblastoma associated with high anti-Hu titres (Wildhaber et al. 2002). Dysautonomia with α3 acetylcholine receptor antibodies is frequently non-paraneoplastic but can occur in association with diverse tumours, most often adenocarcinomas (McKeon et al. 2009) but also thymomas (Rakocevic et al. 2003). CIPO in the context of a thymoma is sometimes associated with myasthenia gravis (Rakocevic et al. 2003; Kulling et al. 1997; Musthafa et al. 2006; Pande and Leis 1999).
Gastrointestinal hypo- or amotility in CIPO is demonstrated by oesophageal manometry, radiological investigations including barium meal examination and Gastrografin enemas (Chinn and Schuffler 1988). CT scan of the abdomen shows bowel distension without mechanical obstruction as does endoscopy of the gastrointestinal tract (Badari et al. 2012). Histopathological examination reveals inflammatory lymphocytic infiltrates in the myenteric plexus associated with Schwann cell proliferation and in most cases neuronal degeneration (Chinn and Schuffler 1988).
Paraneoplastic CIPO represents the minority of all cases with gastrointestinal dysmotility. A frequent cause is progressive systemic sclerosis followed by hollow visceral myopathy (Schuffler et al. 1981). Idiopathic cases of myenteric ganglionitis have been reported (Racalbuto et al. 2008). Familial cases with visceral myopathy associated with CIPO can occur (Sipponen et al. 2009). Amyloidosis may also present as CIPO (Wald et al. 1981). CIPO can develop as a late complication of radiotherapy (Conklin and Anuras 1981) and rarely in Chagas disease (Teixeira et al. 2006).
21.4.7 Lambert-Eaton Myasthenic Syndrome
Lambert-Eaton myasthenic syndrome (LEMS) is another classical PNS of the peripheral nervous systems. Paraneoplastic LEMS occurs approximately fivefold less frequent that SSN and PCD (Giometto et al. 2010). Clinically, it is characterised by predominantly proximal muscle weakness, loss of tendon reflexes, mild to moderate ptosis and autonomic dysfunction, especially dryness of the mouth (O’Neill et al. 1988). In about 10 % of patients with paraneoplastic LEMS, cerebellar ataxia can be found (Titulaer et al. 2011b).
In approximately 50 % of patients, an underlying cancer, almost exclusively SCLC, is detected (Titulaer et al. 2011b). Typically, if present, an initially occult SCLC is diagnosed within the first 2 years after onset by LEMS when thorough tumour screenings including thoracic CT, bronchoscopy and even FDG-PET are applied. In rare cases, prostate cancer, then differentiated in a neuroendocrine phenotype, has been reported associated with LEMS (Tetu et al. 1989; Agarawal et al. 1995). In addition, single patients with thymomas have been reported (Morimoto et al. 2010; Lauritzen et al. 1980).
Antibodies against VGCC are present in 90 % of LEMS (Roberts et al. 1985). However, this finding does not discriminate between paraneoplastic and non-paraneoplastic cases. In approximately 70 % of patients with paraneoplastic LEMS, high titre-antibodies against SOX proteins can be found (Titulaer et al. 2009). Upon immunofluorescence on cerebellar tissue, this antibody corresponds to the anti-glial nuclear antigen (AGNA) pattern (Sabater et al. 2008). Anti-Hu antibodies are found in 30 % of patients with paraneoplastic LEMS (Titulaer et al. 2009). About 10–15 % of patients with LEMS are negative for antibodies against VGCC (Nakao et al. 2002). However, these sera induce similar effects in functional assays. This indicates the presence of antibodies interfering with presynaptic acetylcholine release that are currently undetectable by the available diagnostic tests (Nakao et al. 2002).
Electrophysiological tests can prove the clinically suspected diagnosis of LEMS. Nerve conduction studies show an abnormally low compound muscle action potential (CMAP) in patients with LEMS, which further decreases with low-frequency stimulation (Anonymous 2001). Although this decrement is not specific for LEMS, there is a substantial increment of the CMAP either postexercise or upon high-frequency stimulation (50 Hz) (Hatanaka and Oh 2008).
Using autopsy material, Fukuda et al. could show that in patients with cerebellar ataxia associated with LEMS, there is a marked decrease in cerebellar P/Q-type calcium channel expression (Fukuda et al. 2003).
When the diagnosis of LEMS is established based on clinical and electrophysiological findings, the main differential diagnosis to paraneoplastic LEMS is non-paraneoplastic LEMS. The Dutch-English LEMS Tumor Association Prediction (DELTA-P) score has been reported to accurately predict SCLC in LEMS. This score gives one point each of the following item, if they occur within 3 months after onset of LEMS: age ≥50 years, smoking at diagnosis, weight loss ≥5 %, bulbar involvement, erectile dysfunction in male patients and Karnofsky performance status lower than 70. Whereas a score of 0 and 1 is associated with low risk of an SCLC, a score of 3 and higher is almost invariably associated with paraneoplastic LEMS (Titulaer et al. 2011c). The presence of anti-SOX antibodies has a sensitivity of 67 % and a specificity of 95 % for LEMS with SCLC (Titulaer et al. 2009).
21.4.8 Nonclassical Paraneoplastic Syndromes
Some frequent neurological syndromes can be paraneoplastic in rare occasions. These include brainstem encephalitides, optic neuritis, retinal degeneration, stiff-person syndrome, myelitis including necrotising myelopathy, motor neuron diseases, diverse forms of neuropathy as well as myasthenia gravis and acquired neuromyotonia (Graus et al. 2004) and are described in this section.
Brainstem encephalitis can be observed as part of a generalised paraneoplastic encephalomyelitis but isolated paraneoplastic brainstem encephalitis can also occur in rare cases. Anti-Hu antibodies are associated with a predominantly medullary brainstem dysfunction (Saiz et al. 2009). These patients present with dysarthria, dysphagia and central hypoventilation. Facial, abducens and oculomotor nerve palsies and nystagmus occur frequently, sometimes progressing to complete gaze palsy. In 50 % of the patients, the disease starts with pontine or mesencephalic dysfunction but rapidly descends to the medulla. In contrast, brainstem encephalitis associated with anti-Ma2 antibodies predominantly affects upper brainstem functions. Patients suffer from vertical gaze palsy, and most of these patients have a combination of brainstem, diencephalic and limbic symptoms, sometimes leading to Parkinsonism, excessive daytime sleepiness and chorea (Dalmau et al. 2004). In cases with anti-Ri antibodies and brainstem dysfunction, eye movement disorders, dysphagia and ptosis have been described. Most patients also show postural instability (Sutton et al. 2002). Laryngospasm and/or jaw dystonia has been reported (Pittock et al. 2010). Interestingly, whereas cranial MRI in anti-Hu-associated brainstem encephalitis invariably shows no abnormalities (Saiz et al. 2009), the localisation of clinical brainstem involvement corresponds to T2 or FLAIR hyperintensities in patients with anti-Ma2 antibodies (Dalmau et al. 2004).
Paraneoplastic optic neuritis occurs most frequently in association with anti-CRMP5 antibodies (Cross et al. 2003). These patients also frequently show basal ganglia involvement upon imaging studies and autopsy, which is clinically evident as chorea (Vernino et al. 2002).
Paraneoplastic stiff-person syndrome (SPS) was first described by Moersch and Woltman in 1956. Patients present with chronic fluctuating truncal and proximal muscle rigidity and spasms (Moersch and Woltman 1956). The syndrome is caused by hyperexcitability of spinal motor neurons due to dysregulation of inhibitory GABAergic and glycinergic neurotransmission (Levy et al. 1999; Khasani et al. 2004). The combination of SPS and limbic encephalitis together with prominent myoclonus, hyperekplexia and oculomotor involvement has been referred to as progressive encephalomyelitis with rigidity and myoclonus (PERM) (Meinck et al. 1994). About 80 % of patients with SPS have high titres of anti-GAD antibodies (McKeon et al. 2012). There is an overlap of patients with anti-GAD antibodies and antibodies against the glycine-receptor α1 subunit (McKeon et al. 2013). Only in about 5 % of patients with SPS, a paraneoplastic origin can be assumed due to the coexistence of cancer. SPS is definitely paraneoplastic when anti-amphiphysin antibodies are detected, a finding that is usually associated with breast cancer (McKeon et al. 2012; Murinson and Guarnaccia 2008). Seronegative paraneoplastic SPS have been reported in patients with Hodgkin lymphoma (McKeon et al. 2012). Less well-characterised antibodies associated with SPS include those against gephyrin (Butler et al. 2000) and antibodies against the potassium channel-associated protein DPPX, the latter in patients with a PERM-spectrum syndrome characterised by hyperekplexia and severe diarrhoea (Boronat et al. 2013).
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


