Chapter 6 Parkinson’s disease
Introduction
Idiopathic PD (IPD) accounts for over 70% of all cases of the more general syndrome of parkinsonism (Macphee, 2001). Rare monogenetic variants are important for understanding pathophysiology but account for less than 10% of cases. Secondary parkinsonism may result from a variety of pathological processes including drugs, toxins, trauma and vascular disease. Parkinsonism is a clinical syndrome characterized by slowness of movement (bradykinesia) accompanied by increased muscle tone (rigidity) and resting tremor (Calne et al., 1992). Later in the disease, postural instability and problems with gait, balance and falls become prominent problems (Bloem et al., 2004). Management requires a multi-disciplinary approach with physiotherapists, occupational therapists and speech therapists as vital members in the team (NICE, 2006).
Epidemiology
The incidence of PD in the UK is 18 per 100 000 of population per year, amounting to approximately 10 000 new cases per year. The prevalence of the disease, the total number of cases in the population, is 164 per 100 000 of population. There are approximately 140 000 people with PD in the UK (Meara & Hobson, 2000).
PD becomes more common with increasing age (1% of the population over 65 have PD). Although PD is more likely to occur in males, because females survive longer, there is a fairly even distribution of overall cases between the sexes. PD is age-related with the commonest onset in the seventh decade. Age is the strongest risk factor for developing PD. Onset at a younger age, especially below the age of 35, always raises the possibility of genetic variants of PD (De Lau & Bretele, 2006).
Studies of mortality in PD are limited by the accuracy of death certification and diagnostic confusion between PD and other neurodegenerative conditions. Most studies suggest that PD reduces life expectancy with overall standardized mortality ratios between 1.5 and 2.7 (Louis et al., 1997). Patients with PD are less likely to die of cardiovascular disease or cancer than the general population, but have an increased risk of dying of chest infections (Fall et al., 2003). Cognitive impairment increases mortality significantly (Aarsland, 2008). Patients with PD have a higher risk of hospital and nursing home admissions with consequent ecconomic impact (Vossius et al., 2009).
Aetiology
The causes of sporadic PD are unknown; however, it is likely that the disease results from a combination of factors such as ageing, environmental toxins and genetic susceptibility, resulting in convergent mechanisms causing cell death in vulnerable dopaminergic neurones (Greenamyre & Hastings, 2004).
A variety of environmental factors have been implicated in the development of PD. The most dramatic example is the neurotoxin N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). This substance is chemically related to common pesticides such as paraquat. It was a contaminant of illicit recreational drugs, which led to a mini-epidemic of drug-induced parkinsonism in the early 1980s in California. Patients exposed to MPTP presented with a sudden dramatic onset of PD features and were particularly liable to develop complications such as dyskinesias and cognitive failure (Langston et al., 1983, 1999).
MPTP when given to primates produced a valuable experimental model of PD, although the pathology in the basal ganglia was distinctive, without the characteristic Lewy bodies of the naturally occurring disease. Experiments demonstrate damage to mitochondrial function from free radicals produced as a result of oxidation of MPTP catalysed by the enzyme monoamino oxidase type B. Similar changes in mitochondrial function are present in the natural disease. Some speculate that IPD results from chronic damage to mitochondrial function due to exposure to toxins in the environment, resulting in oxidative stress in dopamine-producing neurones (Schapira, 2008). Potential environmental toxins include pesticides, manganese and copper. Cigarette smoking and drinking large amounts of caffeine have weak protective effects, reducing the risks of PD (De Lau & Bretele, 2006).
Increased familial association in PD was postulated over 100 years ago by Sir William Gowers. Mjones (1949), in a study of Scandinavian families, inferred autosomal dominant inheritance with partial penetration. In a study of a set of twins using the technique of PET scanning, preclinical changes were found which when analysed showed hereditability was 20% (Burn et al., 1992; Ward et al., 1983). The younger the age at onset of the disease, the more likely genetic factors play a role in aetiology.
Genetic studies are reinforcing the impression that PD does not have a single cause. To date, twelve genes associated with rare forms of the disease have been identified. The first Park gene to be discovered (a gene in which an abnormality may cause some cases of PD) was determined in the Italian–American Contursi kindred and was a point mutation in a gene that produces a protein called alpha-synuclein (Polymeropoulos et al., 1996). This protein accumulated in the Lewy body (the hallmark pathological sign of PD). The gene demonstrates autosomal dominance.
A rare form of juvenile PD found in Japan was associated with a mutation in the parkin gene (Lansbury & Brice, 2002). This gene codes for an enzyme associated with the protein ubiquitin found in the Lewy body. Both the abnormalities in the alpha-synuclein and parkin genes affect the ability of the neurone to destroy abnormal proteins. Accumulation of abnormal proteins is a feature of many neurodegenerative conditions, including, most notably, Alzheimer’s disease. The regulation of protein quality within cells by the ubiquitin proteosome system appears compromised in the neurones affected in PD. There are other biochemical abnormalities which indicate that mitochondria are susceptible to damage caused by oxidative stress and that this can trigger cell death. Proteins which are products of other genetic mutations associated with familial PD (PINK1, DJ-1, LAARK2) are allowing scientists to increasingly understand the molecular pathways involved in neurodegeneration and hold out the hope of more effective future treatments (Wood-Kaczmar et al., 2006).
Pathophysiology
The pathology responsible for PD occurs in a group of grey matter structures in the subcortical region of the cerebrum and in the ventral midbrain, the basal ganglia (Flaherty & Grabiel, 1994). They consist of the striatum (caudate nucleus and putamen), the globus pallidus (internal and external parts), the subthalamic nucleus and the substantia nigra (compact and reticular parts). Neurodegeneration occurs most importantly in the pars compacta of the substantia nigra. This area is rich in neuromelanin-containing cells, which give the region its characteristic pigmented appearance. In PD there is less of the pigment as a result of the loss of more than 70% of the neuromelanin-containing neurones. The death of these cells appears to result from programmed cell death (apoptosis) as opposed to necrosis (accidental cell death). Apoptosis is a sequential process initiated by the cell itself in which the genetic material of the cell is enzymatically degraded. Immune cells remove the dying cells. Destruction of this population of cells results in neurochemical changes, the most important of which is dopamine depletion. The substantia nigra is the main source of the neurotransmitter dopamine and projects on to the striatal region. Dopamine is synthesized from the amino acid l-tyrosine. In PD, as the amount of dopamine available falls, compensatory changes occur in the circuitry of the basal ganglia, and these changes are responsible for most of the features we observe in PD.
In the last decade the development of immunostaining for ubiquitin has created a better picture of the distribution of Lewy bodies. Using these techniques, Braak et al. (2006) produced a model of the stages of development of the disease suggesting that the initial pathology is not in the substantia nigra but in the lower brain stem and the olfactory bulb. At the later stages Lewy bodies become plentiful in the cerebral cortex. This model explains many of the clinical features of PD, in particular why patients should have loss of smell as an early feature and why so many patients develop dementia later in the disease (Braak et al., 2006). The extranigral pathology, including non-dopaminergic pathways in the limbic system and cortex, also explains the non-motor features of PD such as autonomic dysfunction, sleep disorders, sensory changes and psychiatric disorders (Lim et al., 2009).
The basal ganglia are part of a series of parallel loops linking with the thalamus and the cerebral cortex (particularly the motor cortex and frontal cortex). Whilst there is considerable debate about the circuitry of the basal ganglia, the classic model proposes two principal pathways concerned with movement – the direct and the indirect pathways (Flaherty & Grabiel, 1994) (Figure 6.1). The direct pathway flows from the putamen and inhibits the internal part of the global pallidus (GPi) and the substantia nigra reticulata (SNr). These two nuclei project to the thalamus. The indirect pathway, as its name suggests, is longer and links the putamen and the external segment of the global pallidus (GPe) via the subthalamic nucleus (STN) to the GPi and the SNr. The two pathways have opposite effects on basal ganglia output to the thalamus. In PD, the decreased production of dopamine leads to an increased inhibitory output to the thalamus so that the rest of the circuit from the thalamus to the cortex suppresses movement, causing bradykinesia. Dopamine is a modulatory neurotransmitter and its deficiency results in a change in background tone, resulting in rigidity and releasing the inhibition of tremor. The medical treatment of PD is concentrated on replacing the deficient dopamine.
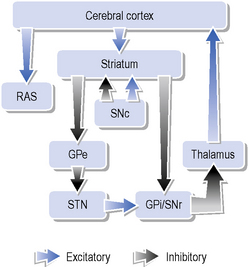
Dopamine is stored in presynaptic vesicles. Action potentials release dopamine across the synaptic cleft. The action on the postsynaptic cell depends on the interaction with dopamine receptors. There are at least six different types of dopamine receptors. These consist of two families, D1-like and D2-like (Strange, 1992). The distribution of dopamine receptors varies throughout the brain depending on the functions undertaken. D2 receptors are most important in mediating motor effects. Both levodopa and dopamine agonists used in the treatment of PD are capable of increasing the stimulation of D2 receptors.
Clinical features
Postural instability, the fourth cardinal sign of PD, develops later in the disease. The characteristic stooped posture is the result of a dominance of flexor tone over extensor tone. Patients may fall backwards (retropulsion) or forwards (propulsion) on examination. Shuffling gait is in part a compensatory change, with the shortening of the stride reducing postural instability. Patients’ ability to initiate or sustain movement may be affected by transient loss of voluntary movement of the feet. Such freezing episodes usually occur late in the disease and are often related to ‘off’ periods, when medication is failing. Freezing and festination (increased step frequency with very small step amplitude), often occur together during specific activities, such as making a turn, or in specific locations, such as narrow spaces (Giladi et al., 2001). Most falls in PD arise from intrinsic (mainly balance disorders) rather than extrinsic (environmental) factors (Bloem et al., 2004).
Non-motor features
The importance of non-motor features of PD which have adverse effects on quality of life is increasingly recognized (NICE, 2006). Mental health problems, difficulties in communication, sleep disorders, falls and autonomic and sensory disturbance (especially pain) comprise the most important of these (Chaudhury et al., 2004). Speech is affected, with the voice becoming monotonous, exhibiting reduced volume and a lack of rhythm and variety of emphasis, with marked psychosocial implications (Miller et al., 2006). Speech impairment is often associated with problems of swallowing. Drooling occurs despite indications of decreased salivary production in PD (Proulx et al., 2005). It is not unusual for patients to complain of muscle pain and this can be due to dystonia, which is an abnormal pattern of muscle contraction, most characteristically toe curling, associated with the wearing off of drug effects.
Autonomic nervous system signs include an increased tendency to bladder hyperreflexia, postural hypotension and sexual dysfunction. Gastro-intestinal manifestations include dysphagia, drooling, weight loss and constipation. Sleep disorders are increasingly recognized as a feature of PD. Daytime hypersomnolence can disrupt rehabilitation. Rapid eye movement sleep disorder, restless legs syndrome, inverted sleep–wake cycle and nocturnal akinesia afflict patients with PD and are often not recognized (Barone et al., 2004). The lack of movement and ability to turn at night can adversely affect sleep (Stack & Ashburn, 2006).
Mental problems complicate the latter stages of PD. Subtle cognitive changes occur early in the disease, particularly frontal lobe executive dysfunction. As the disease develops, other psychiatric problems emerge. Depression, dementia and psychosis are the most common of these (Hindle, 2001). Recognition and active management of these problems is essential as the symptoms can often be improved by drug therapy (Chaudhury & Schapira, 2009).
Making a clinical diagnosis
The diagnosis of PD is based on clinical criteria based on the UK Parkinson’s Disease Society (PDS) Brain Bank Criteria (Gibb & Lees, 1988). Correlation of the clinical diagnosis of PD in practice with postmortem findings in PD brain-bank studies revealed a 25% diagnostic error rate (Hughes et al., 1992a, b). This is very significant as all the patients had been seen by either neurologists or geriatricians who are experts in the field of PD and applied the strictest diagnostic criteria. This research was repeated 10 years later and the error rate had gone down to 10% (Hughes et al., 2001). Nevertheless, in ordinary practice it is likely that some of the patients who have been labelled with PD actually have other related conditions. The commonest differential diagnoses are essential tremor; drug-induced PD; PD plus syndromes such as multiple system atrophy (MSA), progressive supranuclear palsy (PSP); arteriosclerotic parkinsonism; viral infections such as postencephalic parkinsonism; repeated head trauma as experienced by sportsmen such as boxers (dementia pugilistica); rare metabolic diseases such as Wilson’s disease; rare genetic disorders such as Hallervorden–Spatz syndrome; normal-pressure hydrocephalus; diffuse Lewy body disease; cortical basal ganglionic degeneration; and tumours as rare causes of misdiagnosis (Lennox & Lowe, 1997; Litvan, 1997; Quinn, 1989, 1995). The NICE guidelines recommend that anyone suspected to have PD should be referred before treatment has started to a specialist with expertise in the differential diagnosis of the condition and that the diagnosis be regularly reviewed (NICE, 2006).
Therapists must always be alert for atypical cases of PD. In particular, patients who present with symmetrical bilateral signs or who have a disproportionate amount of postural instability at an early stage in the disease should be suspected of having atypical parkinsonism and their diagnosis reviewed. Single photon emission computed tomograhpy (SPECT) can be used to differentiate essential tremor and PD, but imaging is not essential to make the diagnosis of PD (NICE, 2006). Techniques such as positron emission tomography, magnetic resonance spectroscopy and transcranial sonography are used in research and not in routine practice.
Pharmacological management
Two crucial areas of decision-making in relation to medication can be identified:
Patients often need complex combinations of drugs (Bhatia et al., 1998; Olanow & Koller, 1998). Apart from anticholinergic drugs (benzhexol, procyclidine), the symptoms of PD are treated by replacing lost dopaminergic function. The anticholinergic drugs are now only used in younger patients with tremulous disease, as in the longer term they can affect cognitive function and are best avoided in the older patient. They have a range of unpleasant side-effects including dry mouth, postural hypotension and bladder problems, and have low efficacy (Playfer, 2001).
Levodopa (Madopar or Sinemet) is the most widely used drug for Parkinson’s patients. In these drugs levodopa is combined with a decarboxylase inhibitor which prevents peripheral metabolism of levodopa to dopamine. Levodopa is the most effective drug at relieving parkinsonian symptoms; however, its use is associated with two major long-term complications – fluctuations in motor performance and abnormal involuntary movements. Levodopa has a short half-life and its effects can wear off. Once this occurs, increasing dosage of drugs causes involuntary movements affecting the face and tongue or choreoathetoid movements of the limbs. The response to levodopa can become unpredictable and patients can exhibit rapid switches from being ‘on’, where motor performance is well maintained, to being ‘off’, where they are immobile or frozen. Moments of freezing (gait blocks), which are more common in the later stages and after long-term levodopa treatment, can occur in both the ‘on’ and ‘off’ state (Nieuwboer et al., 1997). On–off syndrome becomes so unpredictable that it is extremely disabling to patients and drug strategies are needed to cope with this problem (Clarke & Sampaio, 1997).
The half-life of levodopa can be increased by the use of adjunct therapy. These are enzyme inhibitors which slow the breakdown of levodopa. Catechol-O-methyltransferase (COMT) inhibitors inhibit the enzyme catechol-O-methyltransferase, which is responsible for inactivating levodopa by methylation. One such agent, Entacapone, a reversible peripheral COMT inhibitor, is given together with each dose of levodopa. The drug is well tolerated and has been shown effectively to increase ‘on’ time and reduce wearing-off (Poewe & Granata, 1997). A second agent, tolcapone, has been withdrawn in Europe because of liver toxicity.
Monoamine oxidase type B inhibitors, exemplified by selegiline, inhibit the oxidative metabolism of dopamine. In addition to making levodopa more efficient, selegiline may be neuroprotective. The DATATOP study in the USA showed that use of selegiline before the introduction of levodopa delayed the necessity to start levodopa therapy (Parkinson Study Group, 1989). Although these results were originally interpreted as the drug being neuroprotective, the debate continues. A larger study on this drug, undertaken by the UK Parkinson’s Disease Research Group, indicated that the side-effects of levodopa were increased and that there was increased mortality (Lees, 1995). Since this finding selegiline has been much less widely used. Selegiline has a mild antidepressant and mood-elevating effect. However, it also tends to increase hallucinations and other psychiatric problems.
Many of the dopamine agonists have long half-lives and therefore can approach continuous dopaminergic stimulation. There are currently six available orally acting dopamine agonists and one parenteral drug. Two of these drugs are now not widely used: bromocriptine (because of poor patient tolerance) and lisuride (because of psychiatric side-effects). Pergolide, ropinirole, cabergoline and pramipexole have all been shown to reduce long-term motor complications and are selected for individual patient use on varying pharmacological characteristics (Clarke, 2001).
Apomorphine is possibly the most potent and effective dopamine agonist in the treatment of PD but can only be used parenterally together with a drug blocking its major side-effect of nausea. Apomorphine can be used by means of a pen injection to rescue patients in an ‘off’ state. Continuous subcutaneous infusion of apomorphine has proved an effective treatment for patients with severe motor complications. Use of such drugs depends on availability of PD nurse specialists. A preparation of L-Dopa in a gel (Duo-dopa) that can be infused to achieve continuous dopamine stimulation has recently had success in difficult cases but it is expensive and requires expert support (Poewe, 2009). The dopamine agonist rotigitine is available as a transdermal patch.
The NICE guidelines (2006) conclude that there is no universal first choice drug therapy for either the early or late phases of PD. They also found no strong evidence that any current medication is neuroprotective.
Surgical approaches
With the introduction of levodopa, the need for surgery, widely used prior to the 1970s, diminished but the emergence of long-term complications has led to renewed interest. The success of surgery depends on carefully defined indications and patient selection. Surgery is most suitable for physically fit younger patients who have motor complications refactory to medical treatment but who have previously responded to levodopa and do not have significant mental ill health (NICE, 2006). Two types of surgical approaches are available.
Implantation of fetal cells
Transplanted fetal mesoencephalic cells harvested from aborted fetuses, grown in cell culture and injected into the brain in a form of cell suspension, have been shown to survive in the brain and replace the production of dopamine (Hauser et al., 1999). This type of surgery remains experimental and controversial, and disabling dyskinetic movements following such transplants have been reported in the USA. Fetal transplants were pioneered in Sweden where successful long-term follow-up has been demonstrated (Lindvall, 1998). Manipulation of the patient’s own cells by genetic therapy is an active area of research. The number of transplants undertaken is minuscule compared with the number of patients with PD and the procedure is never likely to be a mass treatment. Cell implants are currently suspended because of complications (severe involuntary movements) associated with the technique (Snyder & Olanow, 2005).
Stereotactic surgery and implantation of stimulators
Better understanding of the pathophysiology of PD has helped to identify areas in the brain that may be targets for stereotactic surgery or the implantation of stimulators. The original target for such surgery was the thalamus. This has been replaced in recent years by targeting either the globus pallidus or, more recently, the STN. The target area of the brain may be lesioned to reduce its output (to benefit the movement disorder) or it may be stimulated (to inhibit output with similar effect). Use of stimulators to the STN can demonstrate dramatic improvements in patients’ motor abilities; however, these procedures are still awaiting the outcome of large prospective double blind controlled trials. Improvement in clinical symptoms after surgery is not reflected in increases in habitual physical activity, and addressing behavioural change through rehabilitation may be warranted (Rochester et al., 2009). Unfortunately, cognitive and psychiatric problems in PD can be worsened by surgery and great caution has to be applied to the selection of patients (Limousin et al., 1998).
Team management of parkinson’s disease
Key priorities for implementation of best practice in PD over the course of the condition are referral to an expert for accurate diagnosis; expert review; regular access to a PD nurse specialist; access to physiotherapy, occupational therapy and speech and language therapy; and consideration of palliative care needs (NICE, 2006). Also relevant to PD service development are the quality requirements, supported by evidence-based markers of good practice, articulated within the National Service Frameworks (NSF), particularly the NSF for Long Term Conditions (LTC) (Department of Health (DH), 2005). The NSF for LTC (DH, 2005) underlines the need for access to the full range of rehabilitation professionals to provide timely, on-going and comprehensive specialist rehabilitation in hospital, in specialist settings and in the community for people with long-term neurological conditions.
The evidence reviewed by NICE (2006) highlighted the key role for the PD nurse specialist in clinical monitoring and medication management and in providing a continuing point of contact for support and information. Occupational therapy was recommended with particular reference to the maintenance of social function and self care, and to environmental and cognitive assessment and intervention. Speech therapy was recognized as valuable in improving quality of speech and communication, including the use of the Lee Silverman Voice Treatment (LSVT). The profession also had a role in the review and management of the safety and efficiency of swallowing and minimizing the risk of aspiration. The NICE (2006) PD guideline reported that there was encouraging high-quality evidence of the effectiveness for some physiotherapy interventions for people with PD. Physiotherapy should be available to people with PD with particular reference to gait re-education, improvement of balance and flexibility; enhancement of aerobic capacity; improvement of movement initiation; improvement of functional independence, including mobility and activities of daily living; and provision of advice regarding safety in the home environment. NICE (2006) also recognized that there was positive evidence of the value of the Alexander technique. A pragmatic phase III randomized controlled trial investigating the clinical and cost effectiveness of physiotherapy and occupational therapy in people with PD (PD REHAB) is currently being undertaken (National Institute for Health Research, 2009). Further trials to investigate the components of physiotherapy (and their effect in the earlier stages of the disease) were recommended (NICE, 2006).
The Primary Care Task Force for the PDS (UK), in their guide for primary care teams (PDS, 1999), highlights the input of the full PD team, including GPs, therapists, social workers, dieticians, as well as experts in neuropsychiatry, neurosurgery and palliative care, in the management of PD in the diagnostic, maintenance, complex and palliative clinical disease stages. However, many patients could not access physiotherapy due to lack of provision (NICE, 2006), a fact underlined by the results of the latest survey of the membership of the PDS in the UK (PDS, 2008), which reported that almost half of respondents had never had a physiotherapy assessment or course of treatment.
Model of physiotherapy management
Morris (2000) underlines the importance of a multi-disciplinary approach and good communication between team members, patients and carers in her seminal model for physical therapy in PD. The basic assumption of the model is that normal movement can be promoted by utilizing treatment strategies that bypass the defective basal ganglia, with the rationale for therapy interventions based on a comprehensive understanding of the pathophysiology and resultant clinical features of the condition (see earlier sections). This knowledge underpins approaches using external (e.g. auditory and visual) and internal (e.g. attentional) cues, which address the size and timing of movements; task-specific training, which addresses difficulties with long sequences of well-learned, automatic movements and performance of multiple tasks; and the environment of therapy to maximize learning, i.e. ideally where the difficulty is most problematic. A sound knowledge of PD medication or surgery and their effect on function is important, together with the effects of ageing, any concurrent pathology, and existing long-term secondary effects of PD and other conditions on the musculo-skeletal and cardiovascular systems (Morris, 2000).
The physiotherapy treatment paradigm is likely to evolve in future to take cognizance of recent work in animal models. Morris (2006) cites work on pharmacological, learning and exercise approaches which point to the possibility of neuroprotection and neuroplasticity in neurodegenerative disorders. The link between rehabilitation and neural adaptation is likely to be an increasing focus of research.





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


