Fig. 9.1
Histology of medulloblastoma (MB) subgroups and Pie chart frequency. (a) Desmoplastic nodular MBs are composed of pale nodular areas with patterns of neuronal differentiation surrounded by densely packed hyperchromatic cells (arrows). These nodular areas produce a dense intercellular fibre network (demoplasia). (b) Medulloblastoma with extensive nodularity shows extensive lobular architecture with large, elongated reticulin-free zones between the nodules (arrows). Internodular zones contain small, round neurocytic-like cells in the fibrillary background. (c) LCA: Large-cell anaplastic medulloblastomas are characterized by enlarged pleomorphic nuclei with prominent nucleoli and variable amount of cytoplasm (arrows). Mitotic and apoptotic figures are abundant. (d) Classic MBs are composed of sheets of small, poorly differentiated cells with arrangements in parallel rows and areas of Homer–Wright neuroblastic rosettes (arrows)
Molecular Heterogeneity
The observation that MB encompasses a number of distinct morphologic variants suggests that these tumors represent different entities arising through alternative mechanisms. Studies of gross chromosomal alterations in MB support this notion and have provided the first clues that different molecular processes underlie the development of MB subtypes (Ellison 2002; Gilbertson 2002; Gilbertson and Ellison 2008).
Deletions of 17p and isochromosome 17q (i17q), which combines loss of 17p and gain of 17q, have long been recognized as the most common chromosomal alterations in MB (Griffin et al. 1988). However, these alterations are not distributed equally among the histologic variants. i17q has been observed in 34 % and 36 % of classic and LCA tumors, respectively, but in only 12 % of desmoplastic MBs (Gilbertson et al. 2001; Lamont et al. 2004; Thompson et al. 2006). Furthermore, the presence of i17q in tumors has been associated with a poor clinical outcome, suggesting that this cytogenetic alteration may contribute to the development of aggressive variants of MB (Batra et al. 1995; Gilbertson et al. 2001).
In contrast, monosomy 6 has recently been shown to occur exclusively in favorable prognosis, mainly classic MBs that contain an intact chromosome 17 and activating mutations in the β-catenin gene (Ellison et al. 2005; Clifford et al. 2006; Thompson et al. 2006) Thus, chromosome 6 may harbor a tumor suppressor gene that cooperates with aberrant Wingless (WNT) signaling to generate an especially curable subtype of MB.
The desmoplastic and LCA variants are also associated with specific chromosomal alterations. Deletions of 9q are observed in up to 40 % of desmoplastic MBs, but occur rarely in tumors of the classic variant (Schofield et al. 1995), and amplifications of the MYCC and MYCN oncogenes occur predominantly in LCA tumors (Ellison 2002). Furthermore, a recent study showed that MB cells transduced with MYCC adopt a severely anaplastic phenotype when grown as xenografts, suggesting a causative relationship between MYCC expression and the LCA phenotype (Stearns et al. 2006).
Aberrations of Genes and Signaling Pathways
Tumor formation is in many ways similar to normal development since both involve processes such as cell proliferation, migration, differentiation and apoptosis. Tumorigenesis is in fact ‘development gone wrong’, whereby developmental signaling or growth factor pathways involved in normal development become aberrant and drive tumor formation. Table 9.1 summarizes signaling and growth factors pathway aberrations.
Table 9.1
Key pathway aberrations and inhibitors associated with medulloblastoma
Pathway | Gene | Aberration | Pathway inhibitor |
|---|---|---|---|
SHH | PTCH | Loss of function | Hh-Antag691 |
SUFU | Loss of function | Cyclopamine | |
SMO | Activation | ||
MYC | Amplification, overexpression | ||
WNT | APC | Loss of function | Unavailable |
β-catenin | Activation | ||
AXIN1 | Loss of function | ||
GSK3-β | Decreased expression | ||
NOTCH | NOTCH2 | Gain | GSI-18 |
HES1 | Overexpression | DFK-167 | |
EGF | ERBB2 | Overexpression | Erlotinib |
MAP2K1 | Activation | ||
MAP2K2 | Activation | ||
MAPK1/3 | Activation | ||
HGF/cMET | cMET | Gain | PHA665752 |
SHH Signaling Pathway
This pathway has been implicated in the pathogenesis of the desmoplastic variant of MB (Behesti and Marino 2009). It plays a major role in proliferation of the GNPCs during cerebellar development. During this time, the SHH glycoprotein is predominantly produced by the Purkinje cells ventral to the EGL and secreted to bind with the PTCH1 receptor on EGL precursor cells. This process releases the PTCH1-mediated inhibition of the SHH pathway, which leads to the activation of target genes via the Gli family of transcription factors and promotes GNPCs proliferation (Fig. 9.2) (Rossi et al. 2008). Consequently, mutations of the SHH pathway may induce aberrant pathway activation and uncontrolled GNPCs proliferation, resulting in the formation of MBs. Mutations affecting PTCH1 and other components of the SHH signaling complex, namely, SUFU, PTCH2 and SMO, have been identified in up to 25 % of sporadic cases of MB (Taylor et al. 2002).
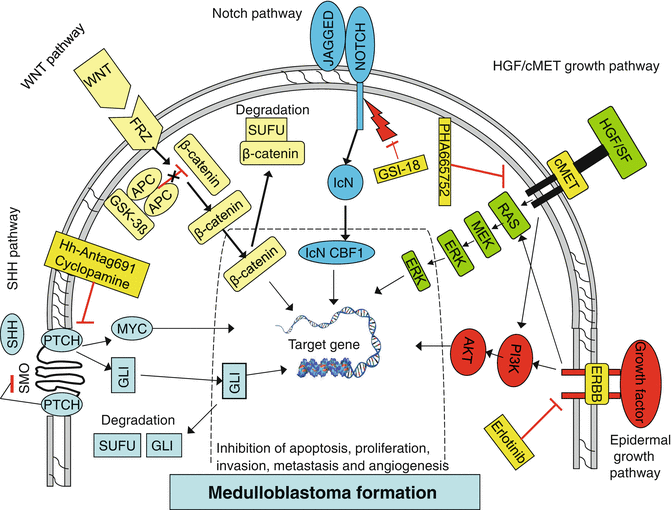
Fig. 9.2
Signaling and growth factor activation pathways implicated in medulloblastoma formation. Arrows activators, bars repressors
Supportive Evidence:
1.
The role of PTCH1 mutations in the genesis of MB has been further supported by murine models, where ~14 % of mice with a heterozygous deletion of Ptc (Ptc+/−) develop MB.
2.
Overexpression of the polycomb transcription regulator BMI-1 has been reported in a subset of human primary MBs and linked with SHH pathway activation. This gene also regulates GNPCs proliferation during cerebellar development and its correlation with SHH pathway activity suggests an alternative mechanism for SHH signaling in the development of MB.
WNT Signaling Pathway
During normal development, WNT ligands bind with the receptor Frizzled (FRZ) to activate this pathway and relay signals to the nucleus via a multiprotein complex (Fig. 9.2) (Rossi et al. 2008). The APC protein is a regulator of WNT signaling that functions in a complex with AXIN, glycogen synthase kinase-binding protein and glycogen synthase kinase-3b to regulate proliferation and specification of neural progenitor cells during early cerebellar development. APC functions as a tumor suppressor by modulating the levels of cytoplasmic b-catenin, a downstream component of the WNT signaling pathway. Inactivating mutations in APC and other WNT signaling cascade members result in an uncontrolled increase in b-catenin levels in the cytoplasm. Consequently, b-catenin translocates to the nucleus where it activates transcription of several oncogenes, including MYC and cyclinD1 (CCND1), resulting in enhanced cellular proliferation.
Supportive Evidence:
1.
Similar observations have also been reported in mutations of SUFU, a downstream component of the SHH pathway, which causes ineffective b-catenin nuclear export resulting in an increased b-catenin-mediated gene expression (Taylor et al. 2004).
2.
Mutations in the WNT signaling complex, especially activating mutations in b-catenin (CTNNB1), account for 15 % of sporadic cases of MBs. In addition, b-catenin mutations and nuclear localization have been associated with a favorable prognosis, but of unknown molecular mechanisms.
NOTCH Signaling Pathway
Notch promotes proliferation of GNPCs and prevents their differentiation (Behesti and Marino 2009). To date, four highly conserved Notch receptors have been identified (Notch1, -2, -3 and -4) that are single-pass transmembrane proteins. Binding of the Notch receptor by its ligand triggers proteolytic cleavage and activation of the receptor, followed by the release of an intracellular domain of the receptor from the membrane that translocates to the nucleus (Carlotti et al. 2008). In the nucleus this protein forms a complex with the DNA-binding protein CBF1, activating the transcription of effector genes including Hes1, Hes5, p21 and cyclin D1 (Fig. 9.2).
Supportive Evidence:
The Notch pathway has been implicated in medulloblastoma tumorigenesis in a number of studies. For example,
1.
Fan et al. 2004 reported increased Notch2 copy number in 15 % of MBs, consistent with its expression in proliferating GNPCs during normal cerebellar development.
2.
Upregulation of the Notch pathway target gene Hes1 in a subset of MB samples.
3.
Hes1 expression also correlated with poor patient survival.
Growth Factors Mutations
In addition to the developmental pathways described earlier, other pathways have been implicated in MB pathogenesis and these have been investigated for a more precise understanding of the molecular basis of this tumor. Abnormalities in the EGF family of receptor tyrosine kinases, for example, have been detected in MBs. Receptor activation through ligand binding leads to its dimerization, autophosphorylation and activation of downstream PI3K and MAPK signaling cascades (Fig. 9.2). This process is essential for normal development of the CNS. Consequently, aberrant activation of this pathway results in upregulation of downstream signaling elements, which leads to increased cell proliferation and altered cell migration through the activation of transcription factor target proteins. In line with this, upregulation of RAS–MAP kinase downstream components, such as MAP2K1, MAP2K2 and MAPK1/3, has been correlated with metastatic behavior of MBs (MacDonald et al. 2001; Del Valle et al. 2002; Gilbertson and Clifford 2003). Furthermore, overexpression of the EGF receptor family member ERBB2 has been reported in MBs and linked to metastasis. High levels of expression of this protein co-overexpressed with ERBB4 signifies poor clinical outcome in cases of MB.
The HGF/cMET signaling pathway is also known to contribute to MB formation (Fig. 9.2). HGF signaling through the cMET receptor plays a critical role in cerebellar GNPCs proliferation and survival as they migrate from the EGL inward to form the mature IGL. Overactivation of this pathway can thus promote malignancies via increased cell proliferation and cell cycle dysregulation and lead to metastatic behavior by abnormal cell migration and invasion.
Aberrant signaling of the HGF/cMET pathway due to loss of pathway inhibition, ligand or receptor overexpression and/or activating mutations has been implicated in several human malignancies, including renal, lung, breast and CNS tumors. In MB, MET gene copy number gains have been detected by comparative genomic hybridization in 38.5 % of primary tumor samples and the receptor mRNA expression levels have been inversely correlated with patient survival(Tong et al. 2004; Li et al. 2005).
Finally, the amplification of MYC and MYCN proto-oncogenes has been detected in 5–15 % of primary MBs (Herms et al. 2000; Aldosari et al. 2002). MYCN is an early transcriptional target of the SHH pathway and its activation by SHH promotes the expression of the cell cycle proteins CyclinD1 and CyclinD2 leading to GNPCs proliferation. A high expression level of MYC is reported to cause progression of MB to an anaplastic phenotype and has been linked to poor clinical outcome.
DNA Repair Pathways and Medulloblastoma
Genes that have recently emerged as important suppressors of MB tumorigenesis are those that regulate the DNA damage response. The DNA repair pathway includes proteins such as poly-(ADP-ribose) polymerase (PARP-1) that sense DNA damage and proteins that repair the damage. DNA double-strand breaks activate two major repair pathways, homologous recombination repair and nonhomologous end joining.
The breast/ovarian cancer susceptibility protein BRCA2 is a DNA repair protein with a key role in homologous recombination repair. BRCA2 colocalizes with Partner and localizer of BRCA2 (PALB2) protein, which stabilizes BRCA2 within nuclear structures, facilitating DNA repair. Proteins involved in nonhomologous end joining include the nuclear ligase Lig4 and XRCC4.
Inactivating mutations in BRCA2 and PALB2 cause Fanconi anemia (FA) types D1 and N, respectively. FA includes a collection of disorders characterized by chromosomal instability, growth retardation, congenital malformations, progressive bone marrow failure, cancer predisposition, and cellular hypersensitivity to DNA cross-linking agents. Mutations in 12 genes have been identified in families with the various forms of the disease. FA-D1 and -N each carry a high risk of childhood solid tumors, including MB. Indeed, at least five of seven childhood brain tumors diagnosed in six FA-D1 kindreds were MBs, and seven patients identified with FAN5 had MB (Offit et al. 2003; Hirsch et al. 2004; Reid et al. 2007).
Although suppression of human MB by other components of the DNA damage response pathway remains to be established, mice null for either Lig4, XRCC4, or Parp-1 and p53 develop MB with high penetrance. Interestingly, most XRCC4-null/p53-deficient MBs delete Ptch1 and, in Parp-1 deficient mice, develop within the EGL. These tumors also express markers of GNPC and Shh pathway activation. Thus, defects in DNA repair may cooperate with mutations in the Shh pathway to cause MB.
Molecular Genetics Analysis of Medulloblastomas
Similar to what happened in WHO Classifications, MB was re-classified more than once depending on its biologic behavior, recent advances in gene expression profiling techniques have led to the generation of several molecular classification schemes in MBs:
1.
 Hyperinsulinemia Tends to Induce Growth Without Growth Hormone in Children with Brain Tumors After Neurosurgery
Hyperinsulinemia Tends to Induce Growth Without Growth Hormone in Children with Brain Tumors After Neurosurgery
 Drugs for Primary Brain Tumors: An Update
Drugs for Primary Brain Tumors: An Update
 Survivors of Childhood Cancer: Risk of New Primary Neoplasms of the CNS
Survivors of Childhood Cancer: Risk of New Primary Neoplasms of the CNS
 Treatment of Brain Tumors: Electrochemotherapy
Treatment of Brain Tumors: Electrochemotherapy
 Brain Metastases: The Application of Stereotactic Radiosurgery and Technological Advances
Brain Metastases: The Application of Stereotactic Radiosurgery and Technological Advances
 Nonthermal Irreversible Electroporation as a Focal Ablation Treatment for Brain Cancer
Nonthermal Irreversible Electroporation as a Focal Ablation Treatment for Brain Cancer




Thompson et al. in 2006 were the first to report molecular subgroups of MBs, identifying five distinct clusters of MB (subgroups A to E). They used a combination of FISH and direct sequencing for gene expression analysis. Subgroup-specific abnormalities included mutations in the Wingless (WNT) pathway and deletion of chromosome 6 (subgroup B) and mutations in the Sonic Hedgehog (SHH) pathway (subgroup D) (Thompson et al. 2006).
Related posts:
Hyperinsulinemia Tends to Induce Growth Without Growth Hormone in Children with Brain Tumors After Neurosurgery
Drugs for Primary Brain Tumors: An Update
Survivors of Childhood Cancer: Risk of New Primary Neoplasms of the CNS
Treatment of Brain Tumors: Electrochemotherapy
Brain Metastases: The Application of Stereotactic Radiosurgery and Technological Advances
Nonthermal Irreversible Electroporation as a Focal Ablation Treatment for Brain Cancer
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
