MAPT (tau)
GRN (progranulin)
Tau
CHMP2B
TARDBP (TDP-43)
FUS
C9ORF72
C9ORF72 is the newest addition to the roster of causative molecules in FTD. In 2011, expansions of a GGGGCC hexanucleotide repeat in C9orf72 were found to cause FTD [1, 2]. Normal alleles usually contain only two of these repeats, and rarely more than 15 repeats [3], while disease-associated alleles contain more than 30 repeats, commonly hundreds or thousands. Patients with C9orf72 mutations can develop behavioral variant FTD (bvFTD), amyotrophic lateral sclerosis (ALS), or FTD-ALS, although other clinical subtypes are also seen and mutation carriers can also develop other neurodegenerative disorders.
Pathophysiology
Three main theories for the pathogenicity of the GGGGCC repeat expansion have been investigated.
First, the expansion is associated with reduced levels of some C9orf72 transcripts [1, 4], suggesting possible loss of function. The repeat expansion is located near the promoter of the gene, and several loss-of-function mechanisms have been proposed. Epigenetic changes, including DNA methylation at CpG sites in the repeat [5] and histone methylation [6], have been reported and could contribute to downregulation of transcription.
Another possibility relates to a unique secondary structure, called a G-quadruplex, formed in guanine-rich nucleic acids, including the GGGGCC repeats in C9orf72 (Figure 15.1A–C). These structures, which are based on hydrogen bonding between guanine nucleotides, occur especially in regulatory regions and may regulate transcription. In C9orf72, the formation of G-quadruplex in the sense DNA strand leaves the antisense strand exposed to more stable interactions with the nascent transcribed RNA [7]. The resulting DNA•RNA hybrid, called an R loop, aborts transcription, resulting in a series of truncated transcripts and reduced levels of the full-length transcript (Figure 15.1D (1)).
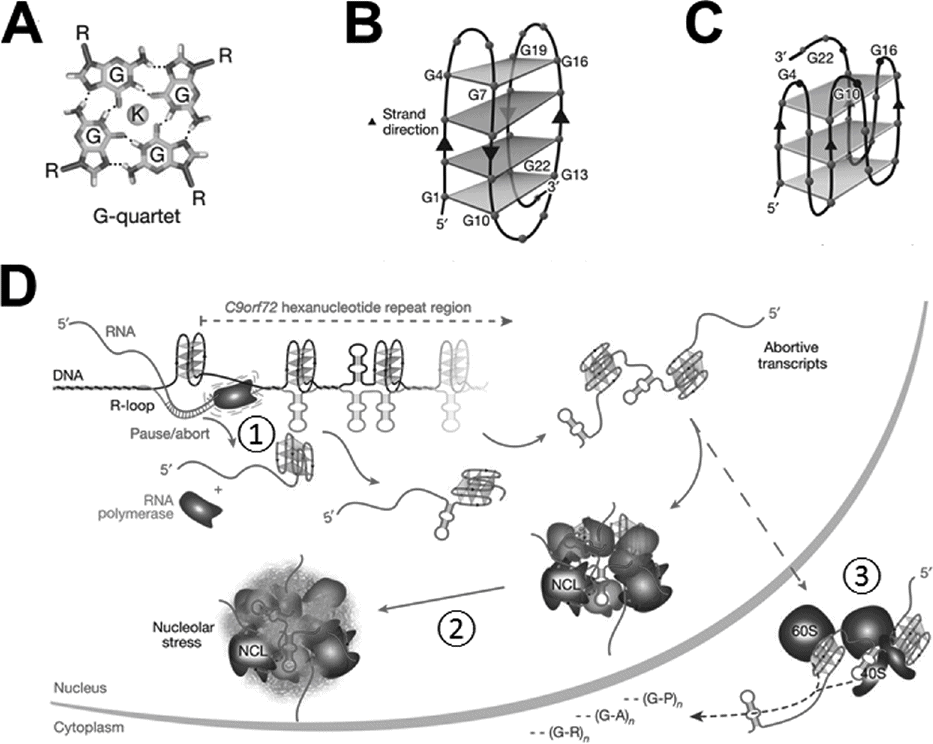
G-quadruplex structures and C9orf72 pathophysiology. (A) Hydrogen bonds between four guanine residues can form a planar structure called a G-quartet. (B,C) G-quartets (gray rectangles) stack together in a G-quadruplex, either with antiparallel strands (B), which may form in GGGGCC DNA, or parallel strands (C), which may form in GGGGCC RNA. (D) Model showing how repeat expansion can be associated with both loss of C9orf72 transcription (1), formation of toxic RNA foci involving nucledin (NCL) (2), and repeat-associated non-ATG (RAN) translation of dipeptide repeats (3).
Second, patients with C9orf72 mutations develop focal RNA aggregates termed RNA foci that may sequester important RNA-binding proteins, causing toxicity due to an RNA gain of function. Similar mechanisms occur in other repeat expansion disorders, including myotonic dystrophy. The GGGGCC repeat has been shown to bind several proteins. In Drosophila, the repeats sequester Pur α, and neurotoxicity can be rescued by overexpressing Pur α [8]. In addition, RNA transcripts containing the GGGGCC repeat also form G-quadruplexes, and these structures bind nucleolin, a major protein in the nucleolus, which is responsible for ribosome assembly and translation [7]. Thus, RNA foci may sequester proteins critical for normal RNA processing and translation (Figure 15.1D (2)).
Finally, the expansion can be translated into an aberrant dipeptide repeat, possibly causing toxic gain of function at the protein level (Figure 15.1D (3)). Another unique cell biologic process appears to be involved, called repeat-associated non-ATG (RAN) translation. As the name implies, RAN translation does not require the typical ATG codon as a translation start site, and leads to formation of homopolymeric (for triplet repeats) or dipeptide repeat peptides (for hexanucleotide repeats). In patients with C9 expansions, there is RAN translation of both sense and antisense transcripts, leading to formation of poly(GP), poly(GA), and poly(GR) from the sense strand and poly(PR) and poly(PA) from the antisense strand [9–11]. Accumulations of these aggregated dipeptide polymers is characteristic in neuropathologic specimens from C9orf72 repeat expansion carriers, although it is not yet clear whether the aggregates are pathogenic or not.
Animal models
A few early animal models of C9ORF72 have been described. In initial approaches to determining whether loss of C9ORF72 has detrimental effects, knockdown of the zebra fish C9ORF72 homologue (zC9orf72), caused axonopathy and swimming deficits [4], but antisense knockdown of C9orf72 in mice had no discernible effect [12]. Meanwhile, expressing GGGGCC(n) repeats in either Drosophila or zebra fish caused toxicity, with RNA foci in the zebra fish model [8, 13]. Thus, the early animal work provides tentative support for both loss-of-function and gain-of-function hypotheses. A host of new models now under development, including transgenic and knockout mice, should help resolve these issues.
Tau
Tau is unique in that it is both a common genetic cause of FTD and a common neuropathologic substrate. The clinical term for cases with tau mutations is frontotemporal dementia and parkinsonism linked to chromosome 17, tau gene (FTDP-17 MAPT). The neuropathologic term is FTLD-tau. These conditions overlap, in that FTDP-17 MAPT patients have FTLD-tau neuropathology [14]. Tau-associated FTD can manifest as many different clinical subtypes, although the semantic variant of primary progressive aphasia (PPA) and FTD-ALS are much less common with tau and are more associated with TDP-43.
Pathophysiology
Mutations in the MAPT gene on chromosome 17q21.1 were the first genetic cause of FTD to be identified [15–17]. Since the first tau mutation discovery, there have been over 40 tau mutations found to cause FTD [18]. Not all FTD-associated tau mutations are alike. Mutations can affect tau at the protein level or pre-mRNA level.
At the protein level, most tau mutations are missense and are clustered in the microtubule-binding domains, which may interfere with interactions with microtubules. A few other missense mutations are outside the microtubule-binding domain at the amino- and carboxy-termini of tau. While tau is mostly unstructured, the termini tend to fold back toward the microtubule-binding domains, which may allow these termini mutations to interfere with microtubule binding. Mutations such as G272V, P301L, V337M, and R406W make tau more prone to hyperphosphorylation [19] and R5L, K257T, I260V, G272V, ΔK280, P301L, P301S, Q336R, V337M, and R406W mutations make tau more prone to aggregation.
At the pre-mRNA level, some missense, silent, or intronic mutations cause alternative splicing of exon 10. Tau can be alternatively spliced to generate six isoforms, including three isoforms that have three microtubule-binding domains (3R) and three isoforms that have four microtubule-binding domains (4R). Inclusion of exon 10 drives formation of 4R tau. Most mutations affecting alternative splicing, such as N279K, L284L, ΔN296, N296N, N296H, P301S, G303V, S305N, S305S, S305I, E10+3, E10+11, E10+12, E10+13, E10+14, E10+16, and E342V, promote the inclusion of exon 10, causing more 4R tau [20]. Other mutations, such as L266V, E9+33, G272V, ΔK280, E10+19, and E10+29, promote exclusion of exon 10, causing more 3R tau [20]. 3R tau has less binding affinity for microtubules and is less efficient at promoting microtubule assembly [21]. Some missense mutations are in exon 10 and therefore only appear if exon 10 is included in the three isoforms of 4R tau.
Tau aggregates in FTLD-tau are made up of post-translationally modified tau. Tau can be modified by hyperphosphorylation, ubiquitination, acetylation, nitration, and glycosylation. Hyperphosphorylation of tau is thought to cause a loss of function as phosphorylation decreases the binding of tau to microtubules, which leads to microtubule destabilization.
The type of FTLD-tau aggregates can sometimes distinguish between clinical syndromes of FTD. 3R tau aggregates (Pick bodies) are found in Pick’s disease (PiD). 4R tau aggregates are found in progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and argyrophilic grain disease (AGD). A mix of 3R and 4R tau is more common and is found in bvFTD and PPA cases.
Animal models
Since FTLD-tau makes up 45% of FTD cases and tau mutations are one of the major genetic causes of FTD, researchers have developed over 25 transgenic mouse models expressing either wild-type or mutant tau to try to recapitulate and understand the pathophysiology of tau.
The first transgenic mouse models expressed wild-type human tau as either the shortest 3R isoform [22] or the longest 4R isoform of tau [23]. In both models, tau was hyperphosphorylated and localized to the somatodendritic compartment, but there were no neurofibrillary tangle (NFT)-like aggregates. Later models increased wild-type human tau expression under different promoters and this led to axonal degeneration [24, 25] and NFT-like aggregates at 18–20 months of age [26], recapitulating some facets of FTLD-tau neuropathology. In another model, overexpression of wild-type tau caused cell loss several months prior to the detection of NFT-like aggregates [27]. These data were the first to suggest that NFT-like aggregates might not be the cause of cell death.
To better understand how tau causes cell death, transgenic mouse models expressing mutant human tau soon followed. Mutant tau (either P301L [28–30] or P301S [31]) was hyperphosphorylated, formed NFT-like aggregates, and caused neuron loss. One of these models, rTg4510 (P301L), further dissociated NFT-like aggregates and cell loss [30, 32]. When the mutant tau transgene was suppressed, NFT-like aggregates continued forming, but cell numbers stabilized and cognition improved. Interestingly, neurons that formed NFT-like aggregates seemed to be less likely to undergo cell death in rTg4510 mice [33]. Additionally, the ultrastructure of cells with NFT-like aggregates appears healthier than that of cells without [34]. On the contrary, a proaggregation mutant tau (ΔK280) causes cell death while mutations that prevent tau aggregation prevent cell death [35]. The most likely hypothesis to explain all of these data is that small, soluble tau oligomers, but not the large, NFT-like aggregates detectable by light microscopy, are responsible for cell death.
Interestingly, functional deficits (synaptic and cognitive deficits) appear independent of NFT-like aggregates and cell death. Neurons with NFT-like aggregates can remain functionally intact [36] and are functionally similar to neurons without aggregates [37]. Synapse loss and cognitive deficits can precede NFT-like aggregates by several months [38]. Most convincingly, in two mouse models where the mutant tau transgene can be turned off, NFT-like aggregates and cell death persist while synaptic and cognitive deficits are reversed [30, 35, 39]. Loss of synapses and/or impaired synaptic transmission coincides with cognitive deficits. In fact, mislocalization of tau to the synapse impairs synaptic transmission before loss of synapses occurs (Figure 15.2A–B) [40] and when synapse loss eventually occurs, it is independent of whether the neurons have NFT-like aggregates (Figure 15.2C) [37]. Research focusing on how tau affects synaptic proteins and synaptic transmission will likely provide potential treatment targets for restoring cognition.
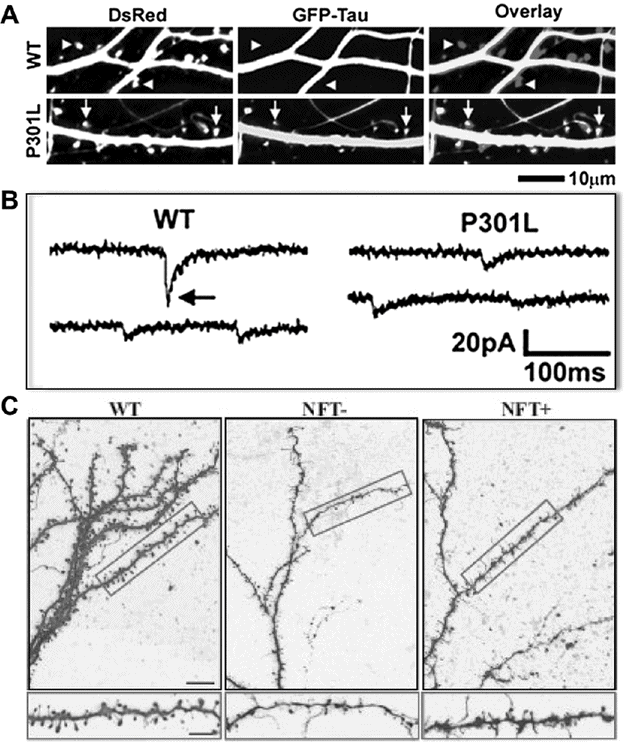
Small, soluble tau, not neurofibrillary tangles, is responsible for neuronal deficits. (A) Dissociated rat hippocampal neurons coexpressing DsRed (to label dendrites and spines) and either wild-type (WT) or mutant (P301L) tau tagged with GFP. Overlap of tau is present in dendrites and spines. In wild-type neurons, most dendritic spines have no tau (triangles), while in neurons from rTg4510 mice, most spines contain mutant tau (arrows). There is no difference in spine density. (B) Representative miniature excitatory postsynaptic current (mEPSC) recordings from neurons in (A). mEPSCs are smaller and less frequent in neurons with P301L mutant tau. Given no difference in spine density, these data suggest mislocalized tau in dendritic spines causes synaptic deficits. (A–B) Reprinted with permission from Hoover BR, Reed MN, Su J, et al. (2010). Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron, 68, 1067–81. (C) Layer III frontal cortex pyramidal neurons from 8.5-month-old rTg4510 (P301L mutant) mice labeled with biocytin and fluorescently stained. Neurons were also co-stained for neurofibrillary tangles (NFTs) with thioflavin-S. Dendritic spine density is decreased in mutant tau mice, but there is no difference between neurons with or without NFTs, suggesting spine loss is independent of NFTs.
Besides impairing synaptic function, tau appears to utilize the synapse to trans-synaptically spread tau pathology to anatomically connected regions. With increasing synaptic activity, tau is released into the extracellular space (Figure 15.3A–B) [41]. Neurons can internalize extracellular tau via bulk endocytosis [42–45], transport tau anterograde and retrograde via endosomal vesicles (Figure 15.3C–D) [45], and spread tau pathology to anatomically connected regions [46–48]. FTD causes region-specific atrophy and reduced connectivity in the salience network [49–52], which consists of functionally and anatomically connected brain regions. The selective vulnerability of these anatomically connected networks is likely due to trans-synaptic spread [53]. Research focusing on how tau is released, endocytosed, and transported could provide potential treatment targets to slow the spread of pathologic tau and, possibly, slow the progression of FTD.
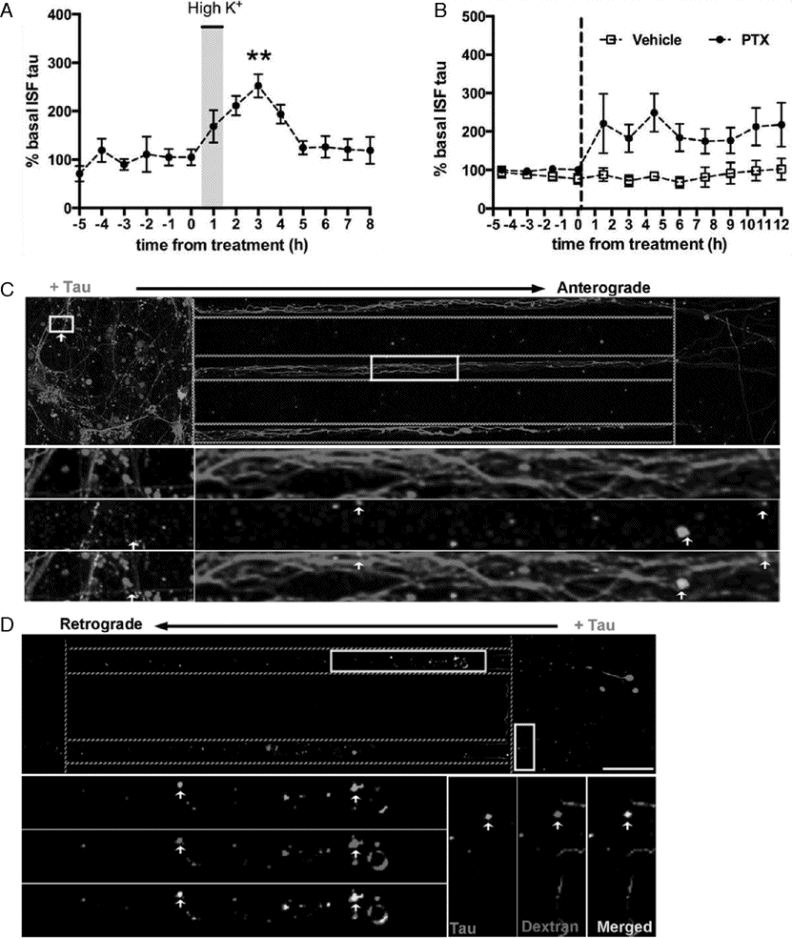
Tau can spread from neuron to neuron. (A) In vivo microdialysis measurements in hippocampus from awake and moving wild-type mice. Increasing neuronal activity by depolarization with high K+ (shaded area) causes increased levels of tau in the interstitial fluid (ISF). (B) Increasing neuronal activity by blocking inhibitory signaling with picrotoxin (PTX) causes increased levels of tau in ISF. (A–B) Reprinted with permission from Yamada K, Holth JK, Liao F, et al. (2014). Neuronal activity regulates extracellular tau in vivo. Journal of Experimental Medicine, 211, 387–93. (C) Microfluidic chambers with cultured neurons compartmentalized into somatodendritic (left), axon shafts (middle), and axon terminals (right). Dotted lines designate chamber separations. Neurons are stained with DAPI (blue) for nuclei and anti-β-tubulin (red) for dendrites and axons. When low-molecular-weight tau (green) is added to the somatodendritic compartment, neurons internalize tau and transport it toward the axon terminals. Also (not shown here), tau co-localizes with markers of endocytosis, dextran, and Rab5, and tau’s uptake is blocked by the endocytosis inhibitor, Dynasore. (D) When tau (green) is added to the axon terminal compartment, tau undergoes endocytosis and retrograde transport down the axon shafts toward the cell bodies. Here, tau co-localizes with dextran (red), a marker of endocytosis. (C–D) Multiple insets show higher magnification of selected areas.
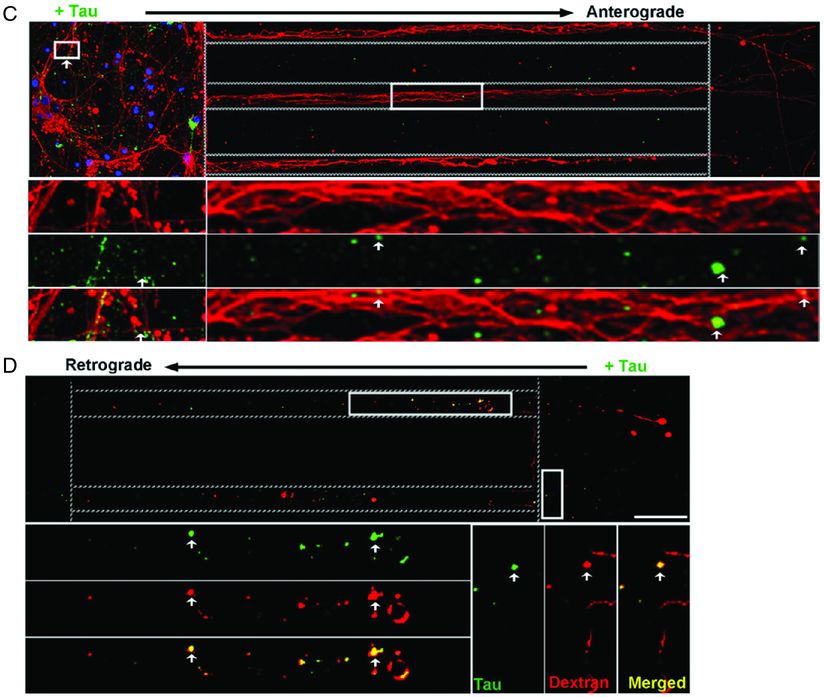
Tau can spread from neuron to neuron. (C) Microfluidic chambers with cultured neurons compartmentalized into somatodendritic (left), axon shafts (middle), and axon terminals (right). Dotted lines designate chamber separations. Neurons are stained with DAPI (blue) for nuclei and anti-β-tubulin (red) for dendrites and axons. When low-molecular-weight tau (green) is added to the somatodendritic compartment, neurons internalize tau and transport it toward the axon terminals. Also (not shown here), tau co-localizes with markers of endocytosis, dextran, and Rab5, and tau’s uptake is blocked by the endocytosis inhibitor, Dynasore. (D) When tau (green) is added to the axon terminal compartment, tau undergoes endocytosis and retrograde transport down the axon shafts toward the cell bodies. Here, tau co-localizes with dextran (red), a marker of endocytosis. (C–D) Multiple insets show higher magnification of selected areas.
Progranulin
Mutations in GRN (the progranulin gene) are one of the three common autosomal dominant causes of FTD. Patients with GRN mutations develop FTLD-TDP (FTLD with TDP-43 proteinopathy) type A, the most common form of FTLD-TDP that is also seen in many sporadic cases [54]. Patients with GRN mutations also rarely present with Alzheimer’s disease clinical phenotypes and amyloid deposition [55].
Pathophysiology
GRN mutations are relatively unique among genetic causes of neurodegenerative disease insofar as they are clearly loss of function, unlike many other genes where it remains unclear whether gain- or loss-of-function effects predominate. Most mutations are either nonsense or frameshift, and some deletions have also been described. These mutations thus cause haploinsufficiency of progranulin. This poses a unique therapeutic opportunity and makes it critical to identify the key progranulin functions that, when lost, cause FTD.
Progranulin is a multifunctional protein, and it is not yet clear which of its functions are most critical in regard to FTD [56]. Progranulin is a secreted glycoprotein and can be cleaved into cysteine-rich granulin peptides. Both progranulin holoprotein and the granulin peptides are biologically active and one issue that is yet to be resolved is whether the effects of GRN mutations are due more to loss of progranulin or granulin activity, or a combination of both.
Progranulin has trophic effects on neurons and serves as a growth factor. Progranulin stimulates neurite outgrowth, and progranulin-deficient neurons have decreased dendritic branching [57, 58]. This function seems to be dependent on a granulin fragment [58]. Progranulin also has trophic effects on other cell types and is overexpressed in some tumors [59].
Progranulin also plays a role in inflammation. The holoprotein has mostly anti-inflammatory effects, while the granulin peptides are pro-inflammatory. It was initially reported that progranulin could compete with tumor necrosis factor-α (TNF-α) for binding to the TNF receptor [60], but others did not reproduce this result [61]. Nevertheless, progranulin knockout mice were more susceptible to inflammatory arthritis [60], and as described below show signs of neuroinflammation. Progranulin-deficient macrophages show increased phagocytosis [62] and an inflammatory profile with increased interleukin (IL)-6 and TNF-α with decreased IL-10 [63].
Finally, progranulin seems to have effects on the lysosome. Homozygous GRN mutations are quite rare, but were reported in one family in which two siblings with undetectable progranulin levels developed a lysosomal storage disorder, neuronal ceroid lipofuscinosis. These individuals developed a syndrome of vision loss, seizures, and cerebellar ataxia in their 20s. This disorder is thus clinically and anatomically quite different from FTD, but the observation suggests that progranulin plays a role in lysosomal function. This is also supported by the development of lipofuscin deposits in progranulin knockout mice [64–66] and genetic interactions between progranulin and the lysosomal gene, TMEM106B [67, 68].
Animal models
Several lines of progranulin knockout (Grn–/–) mice have been described. They model some features of FTD, but not all. Grn–/– mice have neuronal dysfunction with a regional proclivity resembling FTD, with the amygdala among the most affected brain regions [66]. Behaviorally, each of the different progranulin-deficient lines has some abnormalities on social tests [66, 69–71]. Most lines also have some changes in emotional behavior, such as in fear conditioning or anxiety. On the other hand, deficits in hippocampal memory develop only later in life [70, 72].
In terms of neuropathology, Grn–/– mice do not appear to have neuron loss or TDP-43 aggregates. Insofar as these are cardinal neuropathologic features of FTD in patients with GRN mutations, Grn–/– mice are an incomplete model. However, this finding also suggests that loss of progranulin is able to induce neuronal dysfunction without these features. Thus, just as tau pathology is not necessary for the dysfunction induced by tau mutations, TDP pathology is not necessary for the dysfunction induced by progranulin mutations.
Grn–/– mice do have other neuropathologic abnormalities, including microgliosis and astrogliosis [63, 64, 66, 71], and formation of lipofuscin deposits [64–66]. Of course, complete progranulin deficiency in Grn–/– mice more closely mimics the neuronal ceroid lipofuscinosis described above. Interestingly, however, these changes are not seen in heterozygous Grn+/– mice, which model the haploinsufficiency that causes FTD. Grn+/– mice do, though, develop behavioral deficits and amygdala dysfunction just like Grn–/– mice [66]. Thus, the gliosis and lipofuscinosis appear not to be required for neuronal dysfunction due to progranulin haploinsufficiency.
TDP-43 and FUS
Because of several similarities between them, we will consider TDP-43 and FUS together. TDP-43 is one of the common neuropathologic substrates of FTD, including many sporadic cases as well as in patients with C9orf72 or GRN mutations. TDP-43 pathology is particularly common in patients with semantic variant PPA or FTD-ALS. Mutations in the TDP-43 gene, TARDBP, are associated with ALS, but only rarely with pure FTD. FUS pathology is found in about 10% of FTD, making it much less common than tau or TDP-43 pathology. As with TDP-43, mutations in FUS are associated more with ALS.
Pathophysiology
TDP-43 and FUS are RNA-binding proteins with a variety of functions [73]. They are localized predominantly in the nucleus, although they can shuttle back and forth to the cytoplasm. In FTD, both proteins show cytoplasmic mislocalization and aggregation. Thus, multiple models for their toxicity are being examined, including both gain of function (due to either their abnormally high levels in the cytoplasm or toxic properties of their aggregates) and loss of function (either nuclear loss of function due to lower nuclear levels or cytoplasmic loss of function due to abnormal aggregation).
TDP-43 and FUS regulate a variety of RNA-related processes including transcription, RNA splicing, miRNA biogenesis, and translation (Figure 15.4). They are also both components of stress granules [74]. Stress granules are aggregates of RNAs and their associated binding proteins that assemble during times of stress, when it is advantageous for the cell to pause translation of non-essential mRNAs. When TDP-43 and FUS translocate from the nucleus to the cytoplasm, they normally associate with stress granules, then return to the nucleus when the stress resolves and the stress granules dissipate. These dynamics can be disrupted by aberrant aggregation or by impaired nuclear-to-cytoplasmic trafficking of TDP-43 or FUS [74].
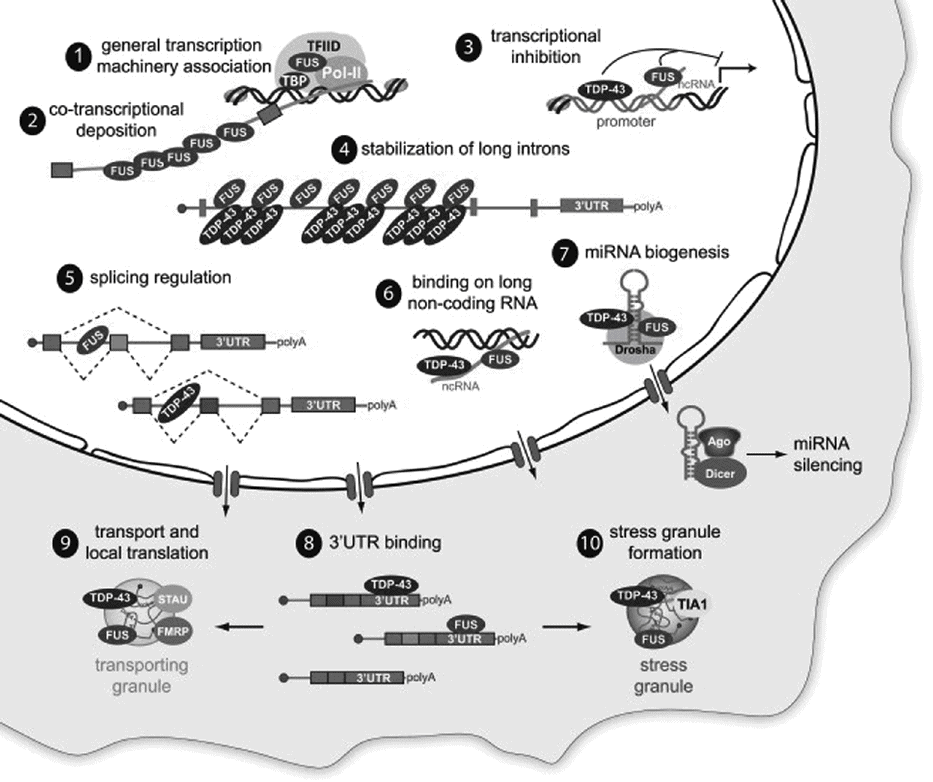
Roles for TDP-43 and FUS in RNA processing and metabolism. TDP-43 and FUS are involved in multiple steps, including regulation of transcription, RNA stabilization and splicing, miRNA biogenesis, RNA transport, and translation control.
Mutations in TDP-43 and FUS more commonly cause ALS, but can be associated with FTD and are instructive for the mechanistic insights they present. Mutations in both TDP-43 and FUS are concentrated in the C-terminal domain [75]. This domain in TDP-43 is important for protein aggregation and has homology to yeast prion-like domains [76]. Mutations in FUS are more distributed, but the majority (and most severe) mutations are in the C-terminus.
Both TDP-43 and FUS contain domains with homology to the yeast prions that contribute to their ability to aggregate [77]. Assembly via these domains is generally reversible and important for their ability to coalesce in stress granules, and thus for their normal physiologic function. However, this potential for prion-like aggregation also confers the potential for spreading of aggregate formation, as discussed above for tau.
Animal models
The first approach to testing for loss-of-function effects is to examine knockout mice. Homozygous Tardbp–/– mice completely lacking TDP-43 were not viable and died at embryonic stages [78–80]. Heterozygous Tardbp+/– mice had normal TDP-43 protein levels and so were not useful for testing loss-of-function effects [78–80]. Postnatal deletion in a conditional knockout led to death within nine days due to increased fat oxidation and resulting weight loss [81]. Most recently, motor neuron-specific conditional knockout mice were found to develop motor deficits and neuronal atrophy [82, 83], consistent with detrimental effects of loss of TDP-43 on neurons.
FUS is similarly embryonic lethal in complete knockout mice [84], but conditional knockouts have not yet been published.
TDP-43 transgenic lines have also been developed to test gain-of-function effects and have been recently reviewed [73, 85, 86]. These efforts demonstrated several salient points, including that both wild-type and mutant TDP-43 overexpression could cause neuronal degeneration. Careful control over TDP-43 levels seems to be critical for neuronal survival, and the gene autoregulates its expression [87]. Several lines had an interesting pattern of selective vulnerability in deep cortical layers, similar to human disease [86]. Cytoplasmic aggregates do not appear critical, as several lines develop motor neuron dysfunction without cytoplasmic accumulation.
FUS transgenic mice have been difficult to generate. Somatic transgenesis using viral vectors to express FUS in the brain yielded mice with some pathologic features of human disease, including cytoplasmic inclusions of aggregated FUS [88]. A transgenic line expressing wild-type FUS develops progressive motor weakness and death within three months due to motor neuron death, with increased cytoplasmic FUS aggregates [89]. Most recently, a line expressing the R521C mutation was described [90]. These mice also die early because of motor neuron loss. Surviving motor neurons had smaller dendritic trees and evidence of DNA damage and RNA splicing defects in genes including that encoding brain-derived neurotrophic factor (BDNF).
VCP
Mutations in VCP are a rare cause of FTD, and there are several atypical features. VCP mutations cause a syndrome of FTD along with bone and muscle disease called inclusion body myopathy with Paget’s disease of bone and frontotemporal dementia (IBMPFD) [91]. Clinically, FTD in IBMPFD can be either bvFTD or semantic variant PPA [92, 93]. Neuropathologically, FTD caused by VCP mutations is associated with FTLD-TDP type D, a distinct subtype that is not seen in sporadic FTD or with other mutations and characterized by TDP-43-positive neuronal intranuclear inclusions and dystrophic neurites with few neuronal cytoplasmic inclusions [94].
Pathophysiology
VCP (called p97 in mouse) is a member of the ATPase associated with diverse cellular activities (AAA+) family. Like other AAA+ proteins, VCP/p97 has numerous functions, including regulating protein degradation through the ubiquitin proteasome system, endoplasmic reticulum-associated degradation, and autophagy. The protein uses its ATPase domain to unfold or refold ubiquitinated proteins and disassemble protein complexes. Disease-associated mutations in VCP are concentrated in a conserved region of the folded protein, and a single mutation, R155C, is responsible in most families. This region of the protein is responsible for recognizing substrates.
Related posts:
 Practical management of frontotemporal dementia
The family’s perspective on FTD
Practical management of frontotemporal dementia
The family’s perspective on FTD
 Neuropathology of frontotemporal dementia and related disorders
Neuropathology of frontotemporal dementia and related disorders
 Functional disability and the impact of frontotemporal dementia in everyday life
Historical introduction to FTD
Functional disability and the impact of frontotemporal dementia in everyday life
Historical introduction to FTD
 Overview of frontotemporal dementia and its relationship to other neurodegenerative disorders
Overview of frontotemporal dementia and its relationship to other neurodegenerative disorders
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


