Pathophysiology of Spinal Cord Injury
Sarah Anne Figley
James W. Austin
James W. Rowland
Michael G. Fehlings
Spinal cord injury (SCI)—which can result from sudden trauma or progressive spinal cord diseases—is a devastating event. Patients suffering from SCI experience significant functional and sensory deficits, as well as emotional, social, and financial burdens. Additionally, individuals with SCI have an increased risk for other health conditions, including cardiovascular complications, deep vein thrombosis, osteoporosis, pressure ulcers, autonomic dysreflexia, and the development of neuropathic pain (1). In North America, it is estimated that approximately 1.5 million individuals are currently living with SCI, with over 12,000 new cases occurring each year (2).
By convention, spinal cord injuries can be divided into two events: a primary and a secondary injury, which refer to the physical injury and the subsequent physiologic cascade, respectively (3). It is well established that pathophysiologic processes that occur in the secondary injury phase are largely responsible for exacerbating the initial damage. These pathologic processes—inflammation, ischemia, lipid peroxidation, production of free radicals, disruption of ion channels, necrosis, and programmed cell death— are rapidly initiated in response to the primary injury and, for the most part, are inhibitory toward endogenous regeneration and repair mechanisms (4,5). The spatial and temporal dynamics of these secondary mediators are fundamental to SCI pathophysiology and are discussed in more detail later in this chapter.
Clinical evaluation and treatment of human patients is complicated by the heterogeneous nature of SCI, which results in variable outcomes for each individual injury. In recognizing the drastic variations of human injuries, the scientific community has been driven to develop and investigate a variety of different animal models. More specific animal models may allow for better translational success, since very few studies that have shown preclinical promise result in similar efficacy upon translation into the clinic.
In the past decade, significant advances have been made, which have notably contributed to understanding the complexity of SCI pathophysiology. However, despite advances in prehospital care, medical and surgical management, and rehabilitation approaches, many patients with acute SCI still experience substantial neurologic disability. Intensive efforts are currently under way to develop effective neuroprotective and neuroregenerative strategies. This chapter aims to summarize the pathophysiologic mechanisms initiated as a result of SCI and, in addition, briefly highlight some potential molecular targets/processes, which may benefit from therapeutic intervention.
PRIMARY INJURY
EPIDEMIOLOGY
The spinal cord is normally protected by the vertebral column; however, physical disruption from trauma often alters the position and structural integrity of the vertebra, leaving the spinal cord vulnerable. Traumatic forces, such as the ones sustained in sports injuries, traffic accidents, and diving into shallow water, cause vertebral column complications associated with SCI (2). Although blunt injury is the predominant cause of SCI, other penetrating trauma due to knife or gunshot wounds occur in a significant percentage of cases (6). Approximately 55% of SCIs occur at the cervical level (C1 to C7-T1), and thoracic (T1 to T11), thoracolumbar (T11-T12 to L1-L2), and lumbosacral (L2 to S5) injuries each account for approximately 15% of SCI (2).
SCI can result from shearing, stretching, laceration, and, in very rare cases, transection of the cord; however, the most common injuries are the result of contusive and compressive forces (2). Worldwide, SCI occurs with an estimated annual incidence of 15 to 40 cases per million. Within the United Sates, new studies suggest that 1.275 million individuals are currently living with SCI—an estimate that is drastically larger than previous estimates of 280,000 individuals—and over 10,000 new injuries occur annually (7,8). Within Canada, it is estimated that 40,000 individuals are currently living with SCI, with approximately 1,000 new injuries occurring each year. Depending on the age of the patient, severity, and level of SCI,
the lifetime cost of health care and other injury-related expenses ranges from $1.25 million to $25 million (9).
the lifetime cost of health care and other injury-related expenses ranges from $1.25 million to $25 million (9).
Perhaps one of the most devastating statistics of SCI is that it predominantly occurs in young, healthy individuals—mostly between 15 and 34 years of age—and statistically, males are almost four times more likely to incur a spinal injury compared to females (10). In the past 30 years, there has been a slight shift in SCI demographics, where the average age of injury has increased from 28.7 to 37.6 years and elderly individuals (those >60 years of age) now account for 10% of SCI cases (an increase from 4.7%) (11). Traumatic SCI can result from a variety of different causes; however, the most common causes of SCI are motor vehicle accidents, falls, work-related injuries, and sports-related injuries (see Fig. 41.1).
ANIMAL MODELS OF SCI
Many small animal models have been developed to investigate the pathophysiology and functional recovery of SCI; however, due to the heterogeneity of the human condition, no one model precisely mimics the complete pathobiologic spectrum. Animal models—usually rats, mice, or other small mammals—include spinal cord compression by forceps or modified aneurysm clips, balloon compression, weight-drop devices/contusion, hemi- or full transection injuries, and chemically induced SCI (12). It has been shown in rats that neurologic impairment increases relative to both the time of spinal compression and the force of trauma (13). Similarly, it has been suggested that reducing the time of spinal compression in human patients, by early surgical intervention and spinal decompression, may result in better functional outcomes for patients (14). Cells of the CNS, particularly neurons and their axons, are prone to shearing and compressive forces, and physica damage results in rapid cell dysfunction and death (15). Although individual animal models fail to address all of the issues present in human SCI, each of the models exhibit at least one of the pathophysiologic consequences. Rat contusion/compression models accurately mimic vascular damage and disruption to the blood-spinal cord barrier (BSCB), cyst or cavity formation, neuronal loss and demyelination, and functional and sensory loss (16,17).
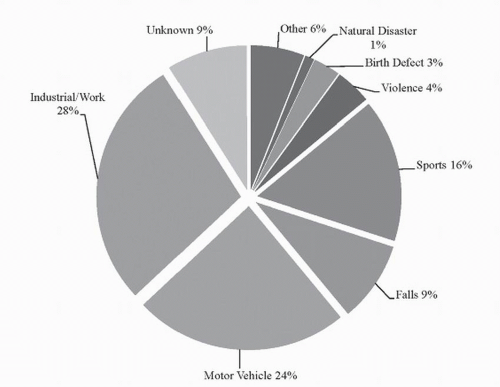 Figure 41.1. Causes of SCI. Graphical representation of 2009 data, which displays the distribution of different causes of SCI in the United States. Adapted from data provided by the Christopher and Dana Reeve Foundation. From One Degree of Separation: Paralysis and Spinal Cord Injury in the United States. (Christopher and Dana Reeve Foundation, 2009) |
MOTOR FUNCTION
A devastating consequence of SCI is paralysis due to damaged axons and neurons in motor pathways at or above the level of injury. Various animal models have been developed to attempt to mimic the motor deficits seen in clinical cases of SCI. Thoracic injuries are well characterized in their functional deficits and result in significant paralysis of the hind limbs. Cervical models of SCI—although not as well characterized as thoracic models—can display decreases in respiratory function and forelimb deficits depending on location and severity of injury. Clinically, spinal cord injuries are divided into one of two general categories: complete or incomplete, referring to total loss of motor and sensory function below the injury site, or partial motor and/or sensory loss, respectively. The American Spinal Injury Association (ASIA) has developed a much more descriptive evaluation, called the ASIA Impairment Scale, to more accurately assess motor and sensory deficits in humans (18).
Motor impairment following SCI is due to both upper and lower motor neuron damage. Loss of lower motor neurons in the anterior horn results in paralysis of muscles at the level of injury. Axons from upper motor neurons, which would normally pass through the injury site (such as ones from the corticospinal tract), are also damaged, and as a result, efferent input to muscles below the level of injury is also impaired (19). Typically, a subpial rim of upper motor axons survives the injury; however, they are left in varying states of demyelination (20). These axons transverse the lesion site and can supply somewhat limited motor information to muscles below—the quality of which is dependent on the number and myelination state of the axons. Since a significant proportion of all injuries occur in the cervical level (-50%), patients experience loss of control to muscles of the upper limbs and diaphragm (in high cervical injuries), and most patients experience some functional deficit in their trunk and lower limbs (10).
SENSORY FUNCTION
Sensory information pertaining to pain and temperature is collected from specialized receptors in the periphery and ascends via the spinothalamic tract to the brain where it is processed (19). First-order neurons entering the CNS must ascend or descend one or two vertebral levels before synapsing on second-order sensory neurons in the posterior horn, which then decussate and continue rostrally to the brain. Conversely, first-order neurons found in the posterior column-medial lemniscus pathway, which transmit fine touch and vibration information, enter the spinal cord and travel rostrally toward the brain prior to decussating in the medulla. Injury to first- and second-order
spinothalamic neurons, or first-order neurons from the medial lemniscus pathway, interrupts sensory information processing at and below the level of injury and prevents normal signal transmission to the brain. Miscommunication in sensory pathways can result in severe complications for patients suffering from SCI. Development of neuropathic pain occurs in many patients, and although the exact mechanism is unknown, it is hypothesized that it is caused by misguided axonal sprouting or abnormal sodium channel excitability in sensory neurons (21). Neuropathic pain is discussed in more detail in the proceeding paragraphs.
spinothalamic neurons, or first-order neurons from the medial lemniscus pathway, interrupts sensory information processing at and below the level of injury and prevents normal signal transmission to the brain. Miscommunication in sensory pathways can result in severe complications for patients suffering from SCI. Development of neuropathic pain occurs in many patients, and although the exact mechanism is unknown, it is hypothesized that it is caused by misguided axonal sprouting or abnormal sodium channel excitability in sensory neurons (21). Neuropathic pain is discussed in more detail in the proceeding paragraphs.
AUTONOMIC FUNCTION
As described above, SCI results in motor or sensory loss as a result of disrupted neural pathways. Unfortunately, damaged neural circuits exhibit more than motor and sensory dysfunction. SCI results in miscommunication between higher centers of the hypothalamus and limbic system, as well as the various effector organs of the autonomic nervous system (22). Preganglionic cell bodies of the sympathetic system are distributed in the intermediolateral horn of the gray matter between T1 and L2, whereas preganglionic cell bodies of the parasympathetic system are situated in the brain stem and sacral levels of the spinal cord. Similar to motor and sensory deficits, the autonomic dysfunction is dependent upon the level of injury. Regulation of cardiac output, vascular tone, and respiration is controlled between T1 and T4; therefore, cervical injuries often exhibit respiratory or cardiovascular complications (19). Gastrointestinal and other organs, including sexual organs, are controlled in lower segments of the spinal cord: T5-L2. In the uninjured spinal cord, spinal sympathetic interneurons remain rather inactive; however, following SCI, they are activated and are thought to contribute to the impaired autonomic function, frequently referred to as autonomic dysreflexia (23).
SECONDARY INJURY
Many factors dictate the pathophysiology of SCI, including the force, severity, and location of the initial injury. Ultimately, these variables are responsible for the extent of molecular damage, tissue loss, and functional deficit. The primary injury, caused by mechanical force, stimulates a complex series of systemic, cellular, and molecular cascades that expand the lesion from the initial injury into surrounding white and gray matter, consequently increasing the extent of tissue loss. Secondary injury is primarily defined by detrimental pathophysiologic responses; however, endogenous repair and regenerative mechanisms are employed during the secondary phase of injury in an attempt to minimize the extent of the lesion and reunite damaged neural circuits.
SUMMARY OF SECONDARY INJURY PROGRESSION
Mammals such as rats and humans (but not mice) develop a fluid-filled cystic cavity at the injury epicenter following traumatic SCI. Over time, this cavity displaces cellular structures as it expands in a rostrocaudal manner from the epicenter, producing significant functional and structural alterations. Infiltrating macrophages, lymphocytes, and activated microglia are present within the cavity, along with granular myelin debris and axons in varying states of demyelination. As mentioned earlier, a subpial rim of tissue often survives the injury and consists of axons that exist in various myelinated states (24). In an endogenous effort to restrict the progression of the cystic cavity, astrocytes are recruited and proliferate, a process termed astrogliosis (25). These astrocytes eventually surround the injury cavity, and in addition to their physical barrier, they express inhibitory molecules, which results in a chemical barrier hindering future attempts of axonal regeneration. Damaged axons undergo Wallerian degeneration toward their cell bodies and away from the epicenter, whereas severed axonal ends distal to the injury site degenerate along with disrupted myelin and are eventually phagocytosed by macrophages (26). The chronic phase of SCI is characterized by the presence of a cystic cavity containing vascular/glial bundles, regenerated nerve roots, collagenous fibers, and astrocytes (27). Refer to Figures 41.2 and 41.3, which provide an overview of pathophysiologic events initiated by traumatic SCI.
THE ACUTELY INJURED SPINAL CORD
Primary physical damage and death of neural cells triggers the acute secondary phase of SCI. Typically, the acute phase represents the first 24 to 48 hours following injury. This phase is characterized by vascular dysfunction, energy and ion imbalances, excitotoxicity, and early inflammatory events that lead to necrotic and, to a lesser extent, apoptotic cell death.
Almost immediately following injury, edema and hemorrhage, which correlate to injury severity, result in ischemic zones and produce necrotic cell death (3,5). Microglia, responding to by-products of necrosis (such as DNA, ATP, K+), become activated and secrete inflammatory cytokines that actively recruit systemic inflammatory cells.
The immediate acute injury is defined as the initial 2 hours postinjury (28). In this phase, spinal shock is initiated below the level of injury, and function is immediately lost (29). Additionally, during this time, neurons and glia that have survived but previously sustained damage from the initial injury hang in the balance of survival and necrotic cell death. Typically, the gross histology of the acutely injured spinal cord has not been significantly altered and may appear normal with MR imaging (30).
Vascular Damage Following SCI
Following injury, drastic changes are observed in the spinal microvascular structure and function (31,32), and histologic studies have shown that the areas with significant vascular damage coincide with the areas of severe neuronal loss (16). Since the gray matter of the spinal cord contains approximately four times more vessels than the white matter, the gray matter is particularly susceptible to vascular injury (33). Vascular changes include reduction in blood flow, hemorrhage, systemic hypotension, loss of microcirculation, disruption of the BSCB, and loss
of structural organization (3,34). Vascular disruption, particularly vasospasm, impaired autoregulation, and loss of microcirculation, is observed almost immediately following injury and has been shown to significantly contribute to the ischemic pathology. Vasospasm occurring after SCI can be initiated by the injury itself or by the release of vasoactive factors (such as nitric oxide [NO] or histamine) (35). Ischemia, resulting from disturbances to the spinal cord blood flow (SCBF), is apparent in almost all cases of human and animal models of SCI, and it has been shown that there is a correlation between injury severity and SCBF (5,36). Following injury, fragile microvascular networks are prone to ischemia since local spinal cord blood pressure is diminished and is further exacerbated by systemic hypotension (37,38).
of structural organization (3,34). Vascular disruption, particularly vasospasm, impaired autoregulation, and loss of microcirculation, is observed almost immediately following injury and has been shown to significantly contribute to the ischemic pathology. Vasospasm occurring after SCI can be initiated by the injury itself or by the release of vasoactive factors (such as nitric oxide [NO] or histamine) (35). Ischemia, resulting from disturbances to the spinal cord blood flow (SCBF), is apparent in almost all cases of human and animal models of SCI, and it has been shown that there is a correlation between injury severity and SCBF (5,36). Following injury, fragile microvascular networks are prone to ischemia since local spinal cord blood pressure is diminished and is further exacerbated by systemic hypotension (37,38).
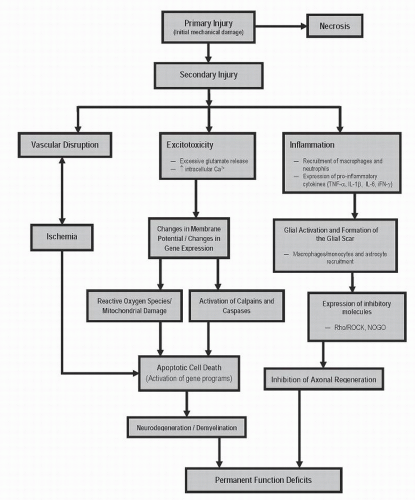 Figure 41.2. A schematic flowchart indicating the progression of traumatic SCI. This diagram summarizes how the primary injury initiates secondary injury cascades, as well as how secondary mechanisms are interconnected. |
Energy, Ion, and Glutamate Imbalances
Dysfunction in metabolic homeostasis, involving imbalances in Na+, K+, Ca2+, and glutamate, is well documented and causes impairment and cell death postinjury. Following injury, axonal concentrations of Na+ and Ca2+ are significantly increased as a result of ion pump failure, inactivation of ion channels, reverse function of ion exchangers, and depolarization of membranes (39,40). Astrocytes and oligodendroctyes also experience increased intracellular levels of Ca2+ following SCI. Through L-type and N-type calcium channels, along with excess glutamate signaling (via metabotropic and ionotropic glutamate receptors), elevated concentrations of Ca2+ may contribute to exacerbation of white matter injury (41,42). Impaired glutamate reuptake by astrocytes is a consequence of glutamate transporter dysfunction, cell death, and glutamate release from glia, neurons, and axons via reversal of Na+-dependent glutamate transport, whereby each event contributes to the excessive extracellular glutamate (41). As early as 3 hours post-SCI, increases in extracellular glutamate is observed, leading to alterations in glial and axonal function and gray matter neuronal cell death (43,44).
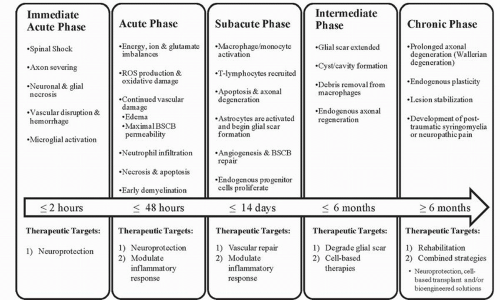 Figure 41.3. An overview outlining the temporal progression of the pathophysiology involved in SCI. This figure describes the timeline of SCI and indicates the key mechanisms that are initiated in each phase of secondary injury. Additionally, this figure highlights potential targets for therapeutic intervention; however, it is not an all-inclusive list. |
Changes in acute energy metabolism following SCI are characterized by diminution of ATP, decreased glucose, and increased lactate/pyruvate ratios (indicative of hypoxia) (45). The ensuing deficits in energy metabolism are undoubtedly due to hypoperfusion/ischemia-mediated decreases in oxygen and loss of glucose availability to cells.
Intracellular Consequences of Acute Excessive Calcium Concentration
Unnecessary accumulation of intracellular Ca2+ results in axonal degradation and neuronal cell death by activation of protein kinases, proteases, and mitochondrial dysfunction. Calpains, a class of Ca2+-dependent proteases, are triggered acutely following SCI due to accrual of intracellular Ca2+. Calpains are known to degrade cytoskeletal proteins, such as neurofilaments and microtubules, which notably disrupt axonal integrity and function (46,47). Additionally, excessive intracellular calcium levels are detrimental to mitochondria, since increased Ca2+ stimulates the production of reactive oxygen species (ROS) in neurons and glia. The production of ROS results in oxidative stress and intensifies neural damage.
Oxidative Stress
As previously mentioned, an increased production of ROS occurs following SCI due to metabolic imbalances and excess intracellular Ca2+, leading to mitochondrial dysfunction and the production of unchecked ROS (48). Experimental methods have determined that peak ROS production occurs 12 hours following injury, with levels remaining elevated until 4 to 5 weeks postinjury, at which point they appear to return to normal levels (49,50). It has been well characterized that ROS initiate necrotic cell death; however, studies have found that brief oxidative stress can cause apoptosis of oligodendrocytes and neurons (51). ROS produced by mitochondria include superoxide (O2–) and hydrogen peroxide (H2O2), and if left unneutralized, O2– freely reacts with NO to produce peroxynitrite (ONOO–), one of the most reactive and detrimental free radicals known. When generation of ROS increases above the antioxidative capacities of cells—as is the case of mitochondrial dysfunction—these reactive molecules can damage proteins, DNA, and lipids. Neutrophil infiltration and rupture have also been recognized as a detrimental source of ROS postinjury through their respiratory (oxidative) burst (52).
Inflammation
Inflammation is a mechanism through which blood-derived leukocytes (neutrophils, monocytes/macrophages, T cells) and soluble factors (cytokines, chemokines, complement, lipid by-products) attempt to restore tissue homeostasis. The inflammatory response initiated following SCI involves a complex interaction between systemic and local
factors. Inflammation postinjury has demonstrated both beneficial and detrimental aspects, including removal of cellular debris and propagation of secondary damage, respectively. Profound differences in inflammatory responses of animal models have been observed between species (even between individual strains of rats). In line with this, inflammatory responses in humans also differ from observations from disease models, rendering direct comparisons between the two difficult (50,53).
factors. Inflammation postinjury has demonstrated both beneficial and detrimental aspects, including removal of cellular debris and propagation of secondary damage, respectively. Profound differences in inflammatory responses of animal models have been observed between species (even between individual strains of rats). In line with this, inflammatory responses in humans also differ from observations from disease models, rendering direct comparisons between the two difficult (50,53).
Within hours of SCI, activated microglia are recruited due to local vascular disruption, loss of tissue homeostasis, and necrotic by-products (ATP, DNA, extracellular K+). In the process of activation, microglia undergo morphologic conversion from ramified to amoeboid and subsequently release cytokines (TNF-a, IFN-gamma, IL-6, IL-1beta) and NO. These cytokines are potent molecules, which recruit systemic inflammatory cells, modulate local protein expression, and result in neurotoxicity and myelin damage (54,55). Neutrophils are the first systemic immune cells to respond to the site of injury, and although they are initially observed within hours after injury, it is not until 24 to 48 hours following injury that they are most abundant (56,70). Neutrophils also secrete matrix metalloproteinases (MMPs), which, upon activation, can cleave endothelial tight junction proteins, leading to increased BSCB permeability. Additionally, neutrophils are a source of myeloperoxidase, which is implicated as an early cause of ROS lipid peroxidation and the subsequent weakening of cell membranes (56). Blocking neutrophil extravasation following experimental SCI has produced both detrimental and beneficial results; thus, it is still debated whether the presence of neutrophils is good or bad following SCI (57,58).
Related posts:
 Developmental Anatomy of the Normal Cervical Spine
Developmental Anatomy of the Normal Cervical Spine
 Pediatric Spinal Cord Injury
Pediatric Spinal Cord Injury
 Calcification of the Ligamentum Flavum of the Cervical Spine
Calcification of the Ligamentum Flavum of the Cervical Spine
 Artificial Disk Surgery of the Cervical Spine: Contraindications for Cervical Disk Arthroplasty
Artificial Disk Surgery of the Cervical Spine: Contraindications for Cervical Disk Arthroplasty
 Controversies, Myths, and Realities Regarding the Surgical Treatment of Cervical Spondylotic Myelopathy with a Special Focus on Laminoplasty
Controversies, Myths, and Realities Regarding the Surgical Treatment of Cervical Spondylotic Myelopathy with a Special Focus on Laminoplasty
 Unilateral Facet Dislocation: Surgery versus Nonoperative Treatment
Unilateral Facet Dislocation: Surgery versus Nonoperative Treatment
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


