Fig. 1
Various OPG presentations. (a) Isolated optic nerve glioma of the left ON. (b) Bilateral ON glioma involving the chiasm. (c) Chiasmal glioma with no involvement of the hypothalamus. (d) Large chiasmal glioma involving the hypothalamus with a cystic component. (e) Large chiasmatic/hypothalamic glioma that compresses the third ventricle, causing hydrocephalus. (f) Posterior glioma of the optic radiations
Several different classification methods have been developed for OPG. Dodge introduced the first method of classification in 1958 [26]. The Dodge system classifies OPGs into 3 classes, based on the anatomical location of the tumor (Fig. 2). Tumors of the ON are termed Dodge I, tumors of the chiasm are Dodge II, and posterior tumors, or those that extend into nearby structures, are Dodge III. This method, defined in the pre-CT and pre-MR era, is still widely used, mainly for research purposes. However, its clinical relevance is limited.
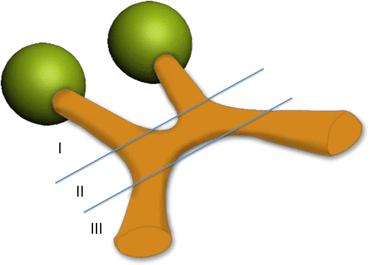
Fig. 2
Schematic representation of the 1958 Dodge classification system. I ON glioma, II Chiasmatic glioma, III posterior glioma or chiasmatic with involvement of extra optic structures
The modified Dodge classification system, developed in 2008 [93], is a more detailed anatomical classification, breaking down each component into several highly precise categories. The modified Dodge also takes into account additional factors such as the existence of NF or leptomeningeal dissemination [93]. This method, while very precise anatomically, is probably too complicated to be routinely implemented in clinical patient care. Another drawback of both Dodge classification methods is the lack of sensitivity to indications of tumor progression.
A third classification system that attempted to address the issue of functional status was suggested by Fred Epstein in 1985 [65]. This classification system is based on an evaluation of two components, tumor/anatomical and functional. The tumor component is rated into one of four classes (T1: one ON, T2: both ONs, T3: OC, and T4: hypothalamus/thalamus). The functional component is rated into one of 5 classes (V0: normal; V1: impaired, one eye; V2: impaired, both eyes, or blind, one eye; V3: blind, one eye and impaired, one eye or field defect; and V4: blind both eyes).
Other factors that should be considered when attempting to stage an OPG patient are the presence of NF1, the age at diagnosis, symptoms at presentation, and the risk of hydrocephalus. We have therefore suggested a new morphologic classification that utilizes recent advances in imaging and may aid in clinical management and patient stratification (Table 1 and Fig 3).
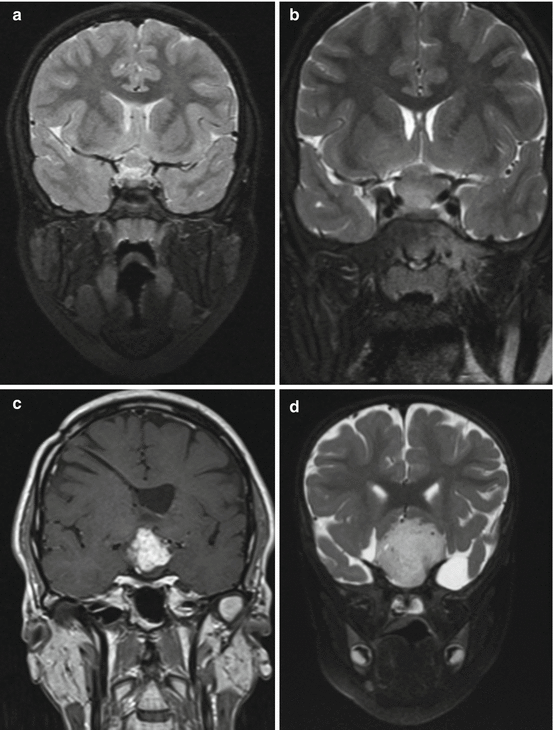
Fig. 3
Four different classes of chiasmatic/hypothalamic glioma. (a) Chiasmal thickening only. (b) Chiasm and hypothalamus. (c) Chiasm and third ventricle involvement. (d) Major suprasellar involvement
Table 1
Our proposed new anatomical classification system for OPG
Nerve | Chiasm | Posterior | General |
|---|---|---|---|
1 – Mid thickening | 1 – Chiasm confined | 1 – Focal involvement | Cyst: |
Yes/no | |||
2 – Severe thickening | 2 – Chiasm and hypothalamus | 2 – Extensive involvement | Hydrocephalus: |
Yes/no | |||
Enlarged ONSD: | 3 – Chiasm and 3rd ventricle | Other CNS | |
Yes/no | Malignancies: | ||
Yes/no | |||
Tortuous ON: | 4 – Major superasellar involvement | Diffuse NF related changes: | |
Yes/no | Yes/no | ||
Pressure on globe: | Age at presentation? | ||
Yes/no | |||
Enhancement: | Enhancement: | Enhancement: | Favorable molecular properties? |
Yes/no | Yes/no | Yes/no | |
Isolated involvement: | Isolated involvement: | Isolated involvement: | |
Yes/no | Yes/no | Yes/no |
In NF1 patients, caution is warranted when attempting to classify OPGs. Despite the fact that this is a relatively common tumor in NF1 patients, there are many radiological abnormalities that may complicate the diagnosis. An example of such misleading abnormalities are T2 hyperintensities that sometimes have mass effect, and focal signal changes within the ON that may either be preneoplastic or without any significance.
Epidemiology
OPGs are relatively rare neoplasms, comprising ~1 % of all CNS neoplasms in the general population and ~5 % of CNS neoplasms in children [84]. The annual incidence (as reported in the pre-MR era) is 1/100,000 [5]. The real number is probably higher. Eighty percent of all diagnosed patients are in the first decade of their lives, and 90 % are in the first two decades. The mean age at diagnosis is 8.8 years in older series, ranging between 2.7 and 5.4 years in more recent series [38, 94, 96]. An estimated 37 % of OPG tend to progress [61]. There is no gender predisposition. In a large series published recently by Nicolin et al., 58 % of 133 OPG patients were NF1 positive. The mean age at diagnosis in this series was 5.9 years, 50 % of the tumors were hypothalamic chiasmatic, 60 % were diagnosed based on imaging only, and 52 % required treatment over the course of a follow-up period of 9 years [68]. The high proportion of Dodge II/III tumors in this series may be attributable to a referral bias.
OPG and NF1
Bilateral OPGs are considered pathognomonic for NF1 and are one of the diagnosis criteria of NF1. The co-occurrence of NF1 in an OPG patient is considered a good prognostic factor [61, 64]. In a recent series comparing NF1 OPG to sporadic gliomas, children with NF1 had a significantly better clinical picture at diagnosis, with less increase in intracranial pressure, less decrease in visual acuity, and fewer abnormalities of fundus of the eye. Radiological progression, visual deterioration, and endocrinological damage were also less frequent in NF1 OPGs.
OPGs appear in almost 30 % of NF1 patients and may present with a variety of radiological and clinical changes [13]. In NF1 patients, the lesion tends to be located anteriorly and involves only a single nerve when compared to generic OPG patients [18]. The spectrum of imaging appearance in NF1 patients is broad, ranging from fine signal changes on T2-weighted MRI, through enlarged optic nerve sheath, up to gross optic nerve tumors and chiasmatic lesions that protrude into the third ventricle or other neighboring structures. When considering posterior tumors in NF1 patients, it is important to remember that they may easily be confused with NF-T2-hyperintensities that may be expansile and have a mass effect. Careful radiological and clinical follow-up is warranted before making any therapeutic decisions for lesions of the optic radiations. Historically, NF1 OPGs were considered relatively indolent tumors that do not tend to progress. Recently, several publications have described a more active clinical course, with progression of the tumor noted in up to 75 % of NF1 patients, even in children older than 11 years old [42]. Recently, macrocephaly was also correlated with OPG existence in NF1 patients [83]. In addition, in a genetic study targeting genotype-phenotype correlation done by Sharif et al., an NF1 patient with OPG was found to have a 6.05 odds ratio to have the NF1 mutation in the 5′ tertile end of the gene when compared to an NF1 patient with no OPG [86].
OPG in Adults
Adult OPGs can be divided into two groups; adult patients with tumors diagnosed in childhood (usually low grade), and adult patients diagnosed during adulthood (usually high grade). In our center, we summarized the data of 22 adult OPG patients. Age distribution at OPG diagnosis varied widely (6 months–66 years), as did age at last follow-up (18–74 years). Ten patients were diagnosed at adulthood, the remaining 12 in childhood.
Of the patients who were diagnosed at childhood (n = 12), 6 had radiological progression during childhood and 3 of those suffered visual impairment. From this group of 6 patients who displayed radiological progression in childhood, one patient had further radiological progression during adulthood accompanied by additional visual decline. Two patients had additional visual decline during adulthood even though there were no signs of radiological progression. The other three patients seemed to stabilize, displaying no additional radiological progression or visual impairment. Of the 6 patients whose tumors were stable or improved (regressed) during childhood, all 6 remained stable with no radiological progression or visual decline in adulthood.
Of the 10 patients diagnosed at adulthood, 6 patients suffered visual deterioration during the follow-up period; in 5 out of these 6 a concomitant radiological progression was noted. Two of the adult-diagnosed patients were diagnosed with high-grade gliomas; both died of their disease.
Eleven patients were diagnosed with NF1, 7 at childhood and 4 at adulthood. Six out of the 7 NF1 patients diagnosed at childhood experienced stable disease with no visual decline, both as children and as adults. Similarly, 3 out of the 4 NF1 patients in the adult-diagnosis group experienced stable disease with no visual decline.
OPGs may be stable or active during childhood or adulthood. As a general rule, patients with radiologically stable tumors diagnosed during childhood did not suffer progression during the follow-up period. Prolonged follow-up should be considered for patients who suffered visual decline or radiological progression during childhood, as well as for late-onset adult OPG patients. OPG is a possible cause for visual decline in adult NF1 patients.
Several practical implications arise from our data. Our experience leads us to recommend prolonged follow-up for pediatric OPG patients who suffered visual decline or radiological progression in childhood, even after a period of stability. In addition, early intervention and more aggressive management may be necessary for adults diagnosed with LGG of the optic pathway. Adults with HGG of the optic pathway demonstrated more favorable prognosis than their counterparts with more commonly located tumors in our limited series.
Clinical Presentation
Visual complaints are the mainstay of clinical symptoms and signs, although may be hard to detect due to the young age of these patients. These include decreased visual acuity (VA), nystagmus, and proptosis, and are found at presentation in 46 % of patients.
Neurological problems, such as headaches, vomiting, and seizures, are present in only 16 % of patients [1]. These low percentages are attributed to the high percentage of patients who are diagnosed during routine screenings, especially in NF1 patients. The diagnostic age for patients with sporadic (non-NF related) OPGs is older than for NF1 patients. This is mainly attributed to the fact that diagnosis occurs only after clinical symptoms appear and not on routine screening, as in the NF1 population [38].
Mode of presentation varies with the anatomical location of the tumor. Posterior tumors are associated with endocrine dysfunction, such as precocious puberty, and with hydrocephalus. Anterior tumors are associated more with visual abnormalities [1]. Visual signs, such as cranial nerve III/IV/VI palsy, papilledema, and optic atrophy, are present in a minority of cases. Visual deficit usually correlates with the tumor location; posterior tumors may cause hemianopia while unilateral ON tumors will cause monocular visual impairment [59]. It is important to remember that assessing visual field in OPG maybe a difficult task due to cooperation difficulties. In our series of 19 patients treated with chemotherapy, visual field was assessable in only 35 % of patients at diagnosis [88]. Signs and symptoms of raised intracranial pressure should be taken seriously, since acute hydrocephalus has been reported as a presenting symptom. Diencephalic syndrome (cachexia, macrocephaly, nystagmus, and visual deficit) is seen in 21 % of infants with chiasmatic/hypothalamic tumors [2].
Diagnostic Studies
Radiology
Today, the gold standard for OPG diagnosis is MRI. To maximize diagnostic yield, MR protocols should be planned with the assistance of an experienced neuroradiologist and should include orbit-directed scanning with fat suppression and contrast injection.
Tractography, the use of diffusion tensor imaging (DTI) MR pulse sequence for the assessment of white matter tracts, is a promising tool [6]. This method was utilized in two studies to assess tumor-induced changes in OPG. In these studies, the OPG caused fibers to either end abruptly within the tumor mass, or induce abnormalities in the arrangement of the visual white matter tracts [29, 62]. Identification of abnormally displaced fibers using this method may also aid in presurgical planning and assist in preserving visual function. The relevance of these changes to the individual patient is still not clear.
Tumors may differ in MR appearance depending on their locations, but they are usually isointense on T1, hyperintense on T2, receive variable enhancement, and have cystic as well as solid nonenhancing components. Tumors of the ON usually do not have cystic changes, appearing as gross thickening of the nerve itself, with or without nerve sheath enlargement. The differential diagnosis for ON enlargement is broad, including meningioma, neuroma, hemangioblastoma, and lymphoma [44]. Optic nerve sheath enlargement is found in nonneoplastic diseases such as increased intracranial pressure, optic neuritis, Grave’s disease, sarcoidosis, toxoplasmosis, central vein occlusion, idiopathic intracranial hypertension, and tuberculosis [76, 87]. Posterior tumors may appear as minimal chiasmal thickening, or as large masses protruding into the third ventricle with apparent mass effect. Cystic changes are common and are a part of the natural history of the tumor. Neoplastic changes posterior to the lateral geniculate nucleus are rare and are difficult to differentiate from NF1 T2 hyperintensities. Unfortunately, initial radiologic appearance does not correlate with visual prognosis. We have found deteriorating vision in patients with tumors that seem to be stable anatomically, as well as stable vision in patients whose tumors demonstrated structural progression. Recently, it has been suggested that dynamic contrast enhancement may correlate with progression, with larger mean permeability values in aggressive tumors [50].
Due to the long follow-up required in these patients and the importance of early detection of changes in the tumor bulk or in its internal components, we recommend the use of volumetric measurements. These measurements, although time-consuming, may improve patient care by enabling more accurate decision making [89, 97]. Figure 4 demonstrates a volumetric follow-up of one of our patients; some of the corresponding MR images are presented in Fig. 5.
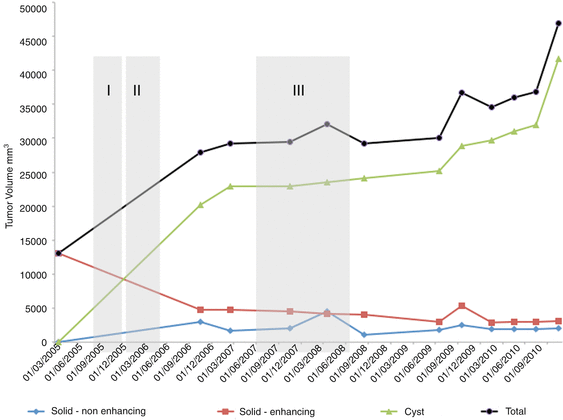
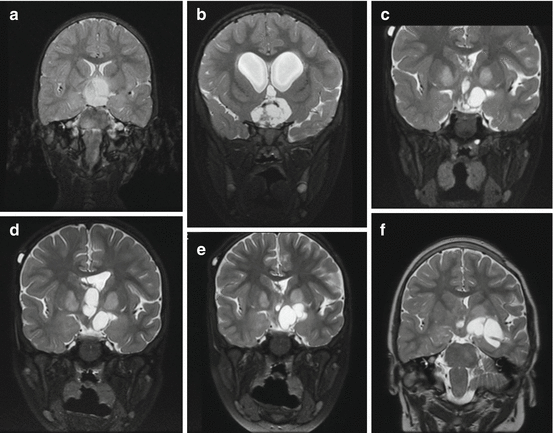
Fig. 4
Volumetric follow-up of one of our patients with internal segmentation into three components and integrated treatment periods. Note that despite the fact that the gross total tumor volume enlarges, there is a marked reduction of the sold component following chemotherapy. The main progression is of the cystic component. Total gross total volume of the tumor, Solid-enhancing volume of the enhancement receiving bulk on MR imaging, Solid-nonenhancing volume of the solid portion of the tumor that does not receive contrast enhancement on MR imaging, Cyst volume of the cystic component on MR imaging, I treatment period with vincristine and carboplatin, II treatment period with vinblastine, III treatment period with rapamycin and tarceva
Fig. 5
Anatomical history of a chiasmatic/hypothalamic glioma. Serial coronal T2 images of a 4-year-old boy with NF1 who presented with visual deterioration [a–f]. Within 4 years, a deterioration to bilateral blindness occurred despite three different chemotherapy treatment lines. Note the cystic changes that the tumor undergoes
Ophthalmology
Neuro-ophthalmology is crucial for both diagnosis and follow-up of OPG, as visual decline is the potentially worrisome consequence of the tumor. Neuro-ophthalmological evaluation is often difficult, especially in young patients where cooperation is limited [8]. A decline in visual acuity is often present at diagnosis and sometimes may be the reason for initial testing, even in the very young (e.g., an infant that starts bumping into objects or sitting closer to the television). In addition, color vision, visual field, eye movements, relative afferent pupillary defect, pupil size, and fundus should all be evaluated. Any progressive change should be considered seriously as a reason to initiate therapy. In very young children, a normal exam does not rule out visual impairment from an OPG. Optical coherence tomography (OCT) has been shown to detect loss of retinal nerve fiber layer in children with OPG (Fig. 6). This may prove to be an auxiliary tool in the diagnosis of visual damage in young children, as well as providing evidence regarding visual reserve and need for treatment [10, 17].
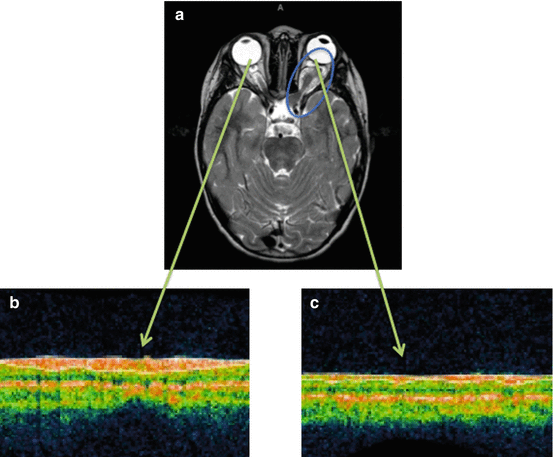
Fig. 6
NF1 OPG patient (a) with an ON tumor on the left nerve (blue circle), Optical Coherence Tomography (OCT) of the right, normal eye (b), and of the left, tumor-affected eye (c) (green arrows). This patient had a VA of 20/20 on the unaffected (right) eye and 20/400 on the tumor-affected eye. VA correlated well with retinal nerve fiber lair thickness of 105 μm on the right (normal) vs. 48 μm on the left (severely thinned) (Images courtesy of Dr. Robert Avery, Children’s National Hospital, Washington DC)
Endocrine
Endocrine assessment should be done in any child with chiasmatic/hypothalamic glioma. Endocrinological abnormalities such as central precocious puberty and growth hormone deficiency are detected in approximately 20 % of OPG patients. Endocrine symptoms may even precede neuro-ophthalmological signs [92]. It should be noted that growth hormone deficiency is present in approximately 2.5 % of NF1 patients even without OPG [19].
Pathology
OPGs are typically low-grade glial neoplasms. Pilocytic astrocytoma (PA) accounts for the vast majority of these tumors. The rest usually consist of fibrillary and pilomyxoid astrocytomas (PMA), oligodendrogliomas, and gangliogliomas. Most of the confirmed pathologies discussed in the literature are usually either PA or PMA. PMA usually present in younger patients, showing a pattern of progression, local recurrence, and seeding through CSF pathways. In a series published by Komotar et al., PMAs were associated with a mortality rate of 33 %, almost twice as much as the numbers for PA (17 %) in this series. In addition, PMAs are also associated with decreased odd survival [3, 52]. A more aggressive behavior pattern may be predicted for labeling indexes such as high ki67 [32, 84] as well as PMA histology [28, 52]. One of the molecular and genetic factors that differentiate PA from PMA is the underexpression of the ALDH1L1 gene, occurring in 89 % of PMA and known to occur in phenotypically aggressive PA [79]. P53 mutations and variable activation of the hedgehog pathway may also be a feature of PMA [3, 81]. Malignant transformation of low-grade OPG is rarely seen; most frequently associated with irradiation [99].
Natural History and Prognosis
OPGs have an erratic natural history. Some of these tumors progress, others are steady for a lifetime, and others spontaneously regress [77, 80]. This has led to the theory that OPGs are not a single entity, but rather a group composed of two or even three subtypes that are often hard to distinguish from one another. These subtypes vary widely in their clinical and radiological outcome. While some OPGs undergo spontaneous regression, there is a significant group of large chiasmatic/hypothalamic tumors that tend to progress and even metastasize. Figure 5 demonstrates a progressive OPG of the chiasmatic/hypothalamic type over 4 years of follow-up.
Interestingly, out of the 5 % of low-grade gliomas that undergo leptomeningeal spread, OPGs represent ~50 % [33]. Before this behavior pattern was recognized, these tumors were sometimes considered benign, even hamartomatous in nature [45], requiring only careful follow-up. OPGs are now perceived as progression-prone, persistent tumors that pose a major therapeutic dilemma. As many as 35 % require treatment at presentation [68]. Patients who do not require treatment at initial presentation have varying chances for progression. Those with NF1 have only 15 % likelihood of progression, while sporadic OPG patients have up to 75 % probability of progression.
Molecular analysis of tumor tissue shows promising early results in predicting the course of the disease. In particular, the BRAF-KIAA 1549 fusion protein seems to indicate tumors with a tendency to arresting growth and even prone to spontaneous senescence. In a recent study, 5-year PFS was 61 % + −8 % for B-K fusion positive patients and 18 % for negative patients. In this study, 61 % of generic OPG were B-K fusion protein positive and no NF1 related OPG was positive for this mutation [41]. Although not currently utilized in routine clinical practice, we believe that this analysis should be considered for any non-NF1 patient in whom therapeutic decisions are made based on radiological progression only.
Treatment
The follow-up and management of OPG requires an experienced multidisciplinary team that provides individualized patient care. Only then can adequate therapeutic decisions be made. The goal of treatment is to prevent visual decline and to achieve long-term tumor control. In the presence of a severe mass-effect or hydrocephalus, immediate, life-saving neurosurgical procedures may be indicated. In most cases, however, OPGs are not a life-threatening tumor. The risk to benefit ratio of treatment must therefore be considered carefully for each patient.
The exact timing of treatment initiation is one of the major open questions in OPG management. In most cases, we recommend delaying treatment for as long as possible unless a clear-cut radiological progression or visual decline is noted. Treating an asymptomatic or minimally symptomatic stable patient seems to offer no advantage over observation alone [7, 32, 96]. Thus, current guidelines suggest intervention only when there is a documented decline in vision or radiological progression [61]. Treatment initiation dilemmas may be very relevant to a child, especially an infant, who presents for the first time with compromised vision. At the moment of presentation, progression cannot be defined. However, compromised reserve may be a relevant reason to start treatment earlier, rather than later.
We recommend imaging and neuro-ophthalmological examinations (including visual field and OCT if available) every 6 months. Patients with compromised reserve for whom a decision was taken not to treat should be clinically examined more frequently. If the tumor is chiasmatic/hypothalamic, a detailed endocrinological evaluation should be performed. If the patient has been stable for a period of a year and has no adverse prognostic factors, follow-ups may be scheduled on a yearly basis.
The choice between treatment options such as surgery, chemotherapy, and irradiation is not easy and depends on the team’s experience and biases. The following sections outline general rules for these treatment modalities. Figure 7 illustrates a suggested management algorithm for OPG.
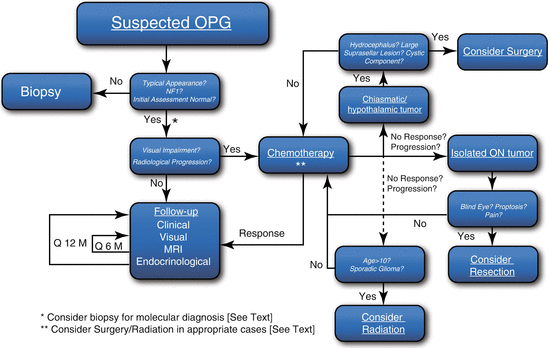
Fig. 7
Management algorithm for OPG patients. ON optic nerve; see text for specifics
Biopsy
For tumors with characteristic MRI appearance that are epicentered on the optic pathway, especially in an NF1 patient, no biopsy is required [60]. Biopsies are indicated if the tumor has an atypical appearance on MR, for an unusual age group (older than 10 years old or younger than 1 year old), or with unusual clinical characteristics (rapidly progressing or severe neurological deficits other than vision loss). When biopsy is indicated, tumor topography dictates the preferred technical method. When the ventricular system is expended, an endoscopic approach can be used to combine multiple procedures, including biopsy, septum pellucidotomy, and accurate shunt placement [21, 22]. Otherwise, a stereotactic needle biopsy can be performed for larger lesions, either with a frame or by frameless techniques [74]. Smaller lesions must occasionally be approached openly through standard microsurgical techniques, or by basal transsphenoidal techniques when the tumor fills the sella turcica. It has been argued that the mere knowledge that an OPG has pilomyxoid characteristics is important for early treatment decisions. The value of a biopsy of an OPG for molecular diagnosis is still controversial. The example of the bRAF mutations, provided previously, may represent only the beginning of a long awaited breakthrough. Biopsies may be useful for molecular diagnosis of tumor characteristics that may have prognostic or therapeutic consequences [41, 75, 95]. These analyses, although not yet included in routine clinical practice, are proving valuable and we expect them to be essential for therapeutic decisions and estimating prognosis in the near future.
Surgery
Clinical series describing surgical results for OPG are usually comprised of small patient numbers, with vague inclusion criteria, no control groups, and poor follow-up. The current situation includes wide variations between different centers and groups with regard to the threshold for open surgery.
Tumors confined to the optic nerve are considered for resection if they show progressive proptosis or intractable pain in a blind eye. These tumors can be approached via unilateral frontal craniotomy and orbitotomy or through the eye, especially when enucleation of the entire eye is warranted. Globe-sparing-resection is also possible. It should be noted that sectioning the nerve close to the chiasm risks damage to the advancing nasal fibers (Wilbrand’s knee) from the contralateral eye.
Surgical management of chiasmatic/hypothalamic tumors is even more controversial. Different microsurgical techniques have been proposed for the removal of large OPG. These different approaches depend primarily on the individual topography of the tumor. Radical resection in a patient with viable eyesight is usually not recommended due to the susceptibility of the surrounding neural structures (hypothalamus and brainstem). In addition, there is a high risk of damage to the optic apparatus and vascular structures [98]. Subtotal resection of tumors that grow outside the visual system, such as the third ventricle, anterior, and lateral subarachnoid spaces, can be performed for a subset of large suprasellar lesions [30]. In addition, subtotal resection may have a beneficial effect on visual outcome via selective decompression of the optic apparatus [90] and may even cause long-term tumor control in selected patients and lessen the hypothalamic dysfunction that is usually associated with more radical resection [43, 98].
Chiasmatic/hypothalamic lesions may be approached via interhemispheric transcallosal, subfrontal pterional, subtemporal, or bifrontal interhemispheric translamina terminalis routes, depending on the direction and the extent of the tumor. Combination approaches may also be used for more extensive lesions. In a surgical series presented by Goodden et al. in 2010, a transcallosal approach was safely used in nine patients with large exophytic tumors that were either causing obstructive hydrocephalus or progressing on serial scanning. Significant debulking (>50 %) was achieved in ~70 % of patients with no incidence of visual deterioration [36]. Image-guidance facilitates accurate navigation and avoidance of damage to critical structures. Hydrocephalus may respond to decompression alone, but in many cases may also require shunt placement, which is usually the preferred method [82]. Ascites secondary to ventriculoperitoneal shunting in chiasmatic tumors is common and may require diverting CSF to the atrium rather than to the abdomen [35]. Large cystic components containing mucinous fluid are common and may require multiple resections and drainages. Shunts placed within these cysts tend to malfunction after a short period of time; Ommaya reservoirs may be used in these situations for intermittent tapping and drainage.
Chemotherapy
Chemotherapy is considered the first line of treatment in most cases of children with progressive OPG.
“Gentle Chemotherapy” with vincristine and carboplatin was introduced in 1987 by Packer et al. and is now an accepted first-line treatment [71]. This regimen reported PFS rates of 75 % at 2 years and 68 % at 3 years for chiasmatic tumors [71]. Carboplatin alone may also be effective in OPG treatment, with a short-term PFS rate of 83 % and disease stabilization in 85 % [4]. Weekly vinblastine is also frequently used as first line or in patients with carboplatin allergies [58]. Recently, a new protocol of bevacizumab and irinotecan has shown preliminary effectiveness in the treatment of recurrent low-grade gliomas [72]. Temozolomide was suggested as a possible option for second- or third-line treatment with an overall disease stabilization rate of 54 % in patients with progressive OPG [40].
Adverse effects of chemotherapy vary according the regimen and drugs used. Among the side effects of the vincristine and carboplatin protocol are carboplatin-associated allergies occurring in ~10 % of treated patients [73], and peripheral neuropathy. Hematological side effects such as neutropenia and thrombocytopenia may occur with this protocol, but are usually associated with nitrosourea-based regimens [78]. Temozolomide was shown to cause grade 2–4 thrombocytopenia and neutropenia in ~23 % of treated pediatric OPG patients in addition to intratumor hemorrhage in ~3 % [40].
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


