CHAPTER 21
Pediatric Neurology
I. Embryology and Development
A. Embryology of nervous system and corresponding disorders (high-yield items)
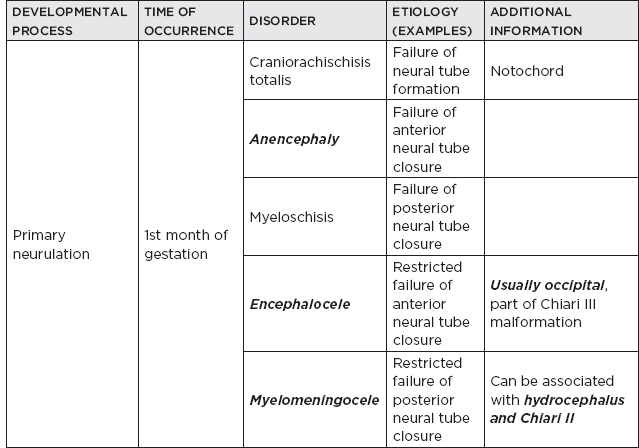
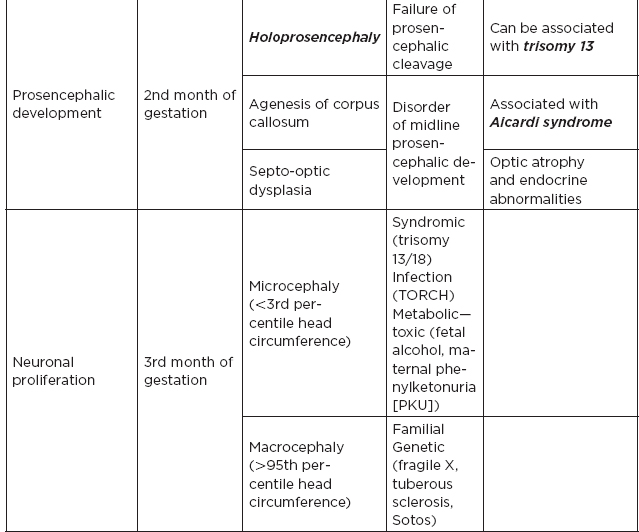
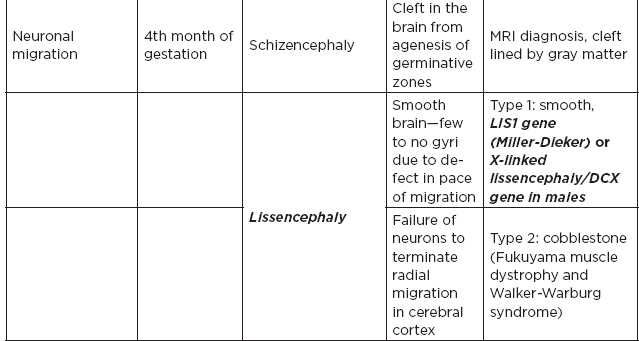
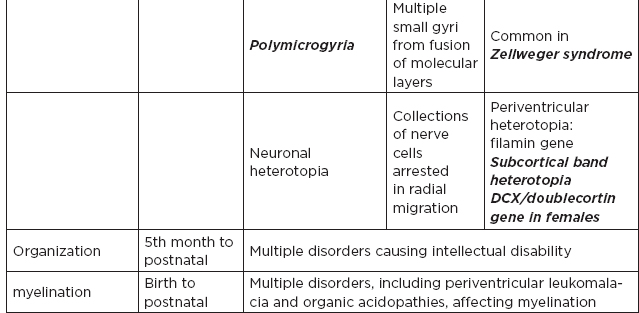
1. Risk factors for neural tube defects: maternal diabetes (caudal regression syndrome), folate deficiency and use of antiepileptics during pregnancy (especially valproic acid)
2. Occult dysraphic states: High suspicion with abnormal tuft of hair, skin dimples or tracts
a. Myelocystocele—localized cystic dilation of caudal spinal cord
b. Diastematomyelia—bifid spinal cord
c. Meningocele—no spinal cord tissue in sac, usually contiguous with tumors; lipoma, teratoma
d. Tethered cord—caudal end of the spinal cord fixed with fibrous bands
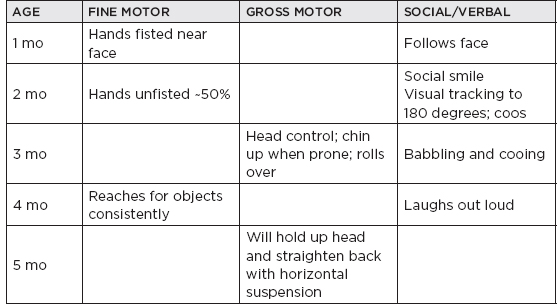
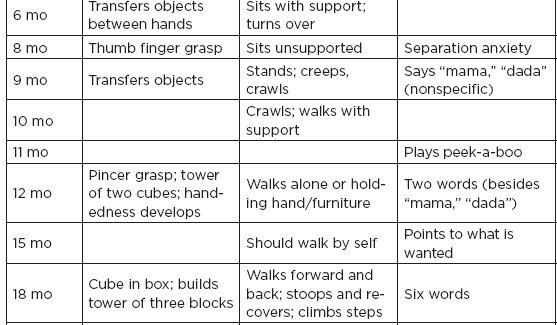
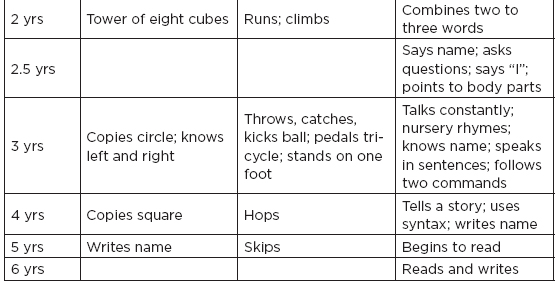
B. Normal developmental milestones
II. Neonatal Neurology
A. The neonate nervous system functions essentially at a brainstem–spinal level; examination should be directed to diencephalic–midbrain, cerebellar–lower brainstem, and spinal functions; control of respiration and body temperature, regulation of thirst, fluid-balance appetite (hypothalamus and brainstem); automatisms, sucking, rooting, swallowing, grasping (brainstem–cerebellum); movements and postures of neck, extension of neck, trunk, flexion movement, steppage (reticulospinal, cerebellar, spinal); muscle tone of limbs and trunk; reflex eye movements (tegmental midbrain, pons); state of alertness (diencephalon); reflexes: Moro.
B. Intraventricular hemorrhage (IVH): common in preterm infants, especially very-low-birth-weight infants (<1,500 g); originates from rupture of germinal matrix vessels; screening and monitoring done with serial cranial ultrasound; increased incidence of hydrocephalus with Grade III to IV IVH
IVH GRADING SYSTEM | |
I | Germinal matrix hemorrhage |
II | Intraventricular hemorrhage with NO ventricular dilatation or hemorrhage <50% of ventricles |
III | Intraventricular hemorrhage WITH ventricular dilatation or hemorrhage >50% of ventricles |
IV | Intraventricular hemorrhage with hemorrhagic infarction into the parenchyma |
C. Periventricular leukomalacia (PVL): white-matter injury with cystic changes usually associated with high-grade IVH or decreased cerebral blood flow in watershed areas
D. Hypoxic-ischemic encephalopathy (HIE): “neonatal encephalopathy”; may be due to prenatal (maternal/placental disease), perinatal (difficult delivery), or postnatal factors (trauma), usually presents as neonatal seizures
1. Two patterns: (1) acute severe asphyxia leads to damage in deep gray-matter areas; (2) partial prolonged asphyxia (more common) leads to cortical involvement with edema and watershed injuries.
2. Can be mild, moderate, or severe and be associated with multiple-organ involvement. Moderate to severe HIE can be treated with hypothermia (whole-body or head cooling) within the first 6 hours.
E. Cerebral palsy (perinatal encephalopathy): defined as a fixed, nonprogressive neurologic motor deficit of multiple etiologies; not necessarily cognitive impairment
SUBTYPE | MAIN INJURY | CLINICAL FEATURES |
Spastic diplegia (most common) | Preterm: PVL | Spasticity in lower extremities Can have normal intelligence |
Spastic quadriplegia | Preterm: PVL Term: HIE and central nervous system (CNS) infections | Bilateral spasticity Epilepsy and cognitive impairment common |
Spastic hemiplegia | Preterm and term: congenital malformations, perinatal stroke | Early handedness Epilepsy common 1/3 with normal intelligence |
Dyskinetic (extrapyramidal or athetoid) | Usually term infants: basal ganglia injury—kernicterus | Dystonic movements Normal to borderline intelligence |
Ataxic, mixed types (less common) | Varied, may be genetic syndromes or congenital malformations |
|
F. Floppy infant
1. Cerebral lesion: atonic cerebral palsy, Prader-Willi, Down syndrome, storage/amino acid disorders
2. Cord lesion: transection during breech delivery, myelopathy from umbilical artery catheters, spina bifida, dysraphism
3. Anterior horn cell: spinal muscular atrophy (Werdnig-Hoffman, Kugelberger-Welander) Pompe’s, poliomyelitis
4. Peripheral nerves: rare, congenital peripheral neuropathies
5. Neuromuscular junction: botulism, aminoglycosides, hypermagnesemia (from maternal treatment of eclampsia)
6. Muscle: nemaline rod, central core, myotubular myopathy, congenital muscular dystrophy
7. Systemic: hypercalcemia, hypothyroidism, renal acidosis, celiac, cystic fibrosis, Marfan, Ehlers-Danlos
8. Benign: Amyotonia congenita (diagnosis of exclusion)
III. Inherited Metabolic Disease of the Nervous System: The nervous system is the most frequently affected system by genetic abnormality; one-third of all inherited diseases are neurologic.
A. Modes of inheritance
1. Autosomal dominant (AD): manifest disease as heterozygotes, but variation in the size of the gene abnormality; may produce several phenotypes; variable degree of penetrance and expressivity are characteristic; tendency to appear long after birth
2. Autosomal recessive (AR): more uniform phenotypic expression, onset soon after birth, usually an enzyme deficiency
3. X-linked: mutant gene affects mainly one sex; Lyon hypothesis: female will experience same fate as the male if one X chromosome is inactivated in most cells during embryonic development; biochemical abnormality more often a basic protein.
4. Multifactorial genetic disease: may present as constitutional disorders with gene abnormalities located on several chromosomes (polygenic); relative contributions of “risk genes” and environmental influences are highly variable.
5. Mitochondrial disease: mitochondrial DNA: double-stranded circular molecule that encodes protein subunits required; essential feature: inherited maternally; genetic error is most often single-point mutation; may also be deletions or duplications that do not conform with maternal inheritance (sporadic, e.g., Kearns-Sayre); some enzymes of respiratory chain are coded by nuclear DNA, which is imported to the mitochondria, resulting in a Mendelian pattern of inheritance.
B. Metabolic disorders
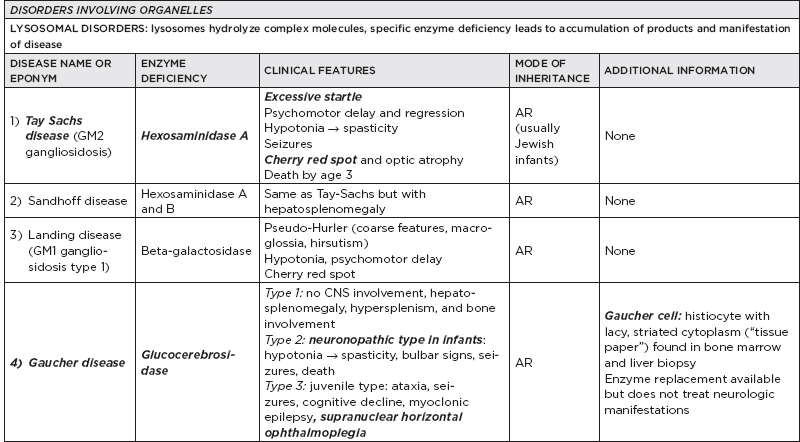
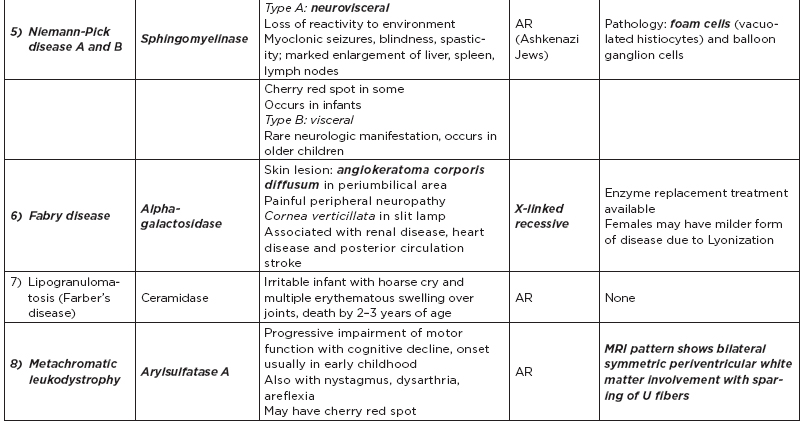
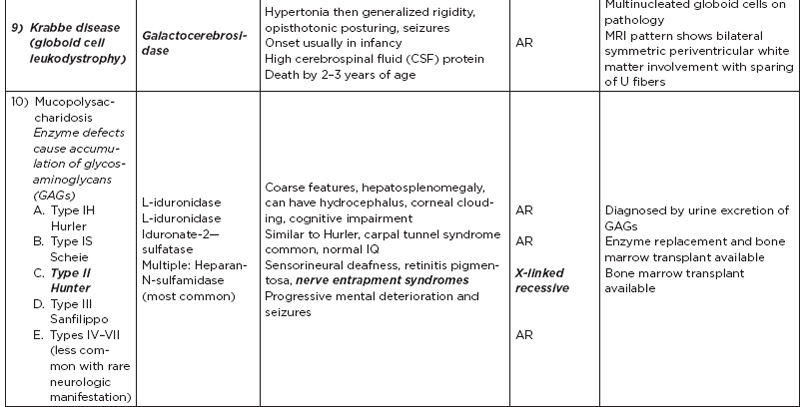
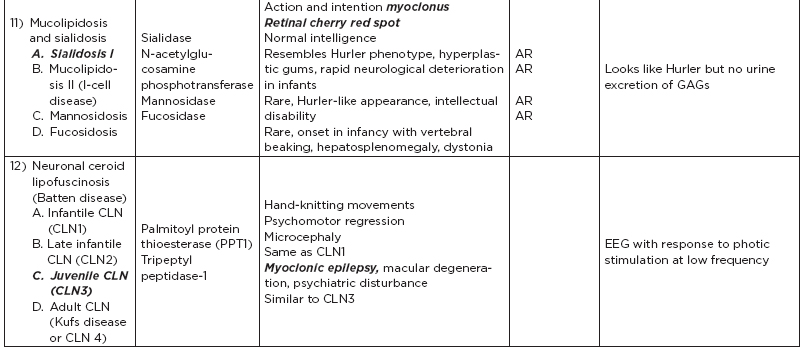
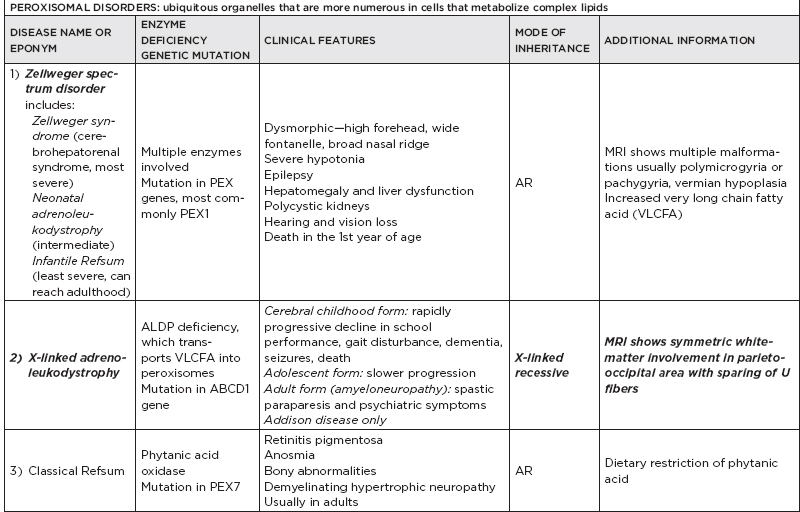
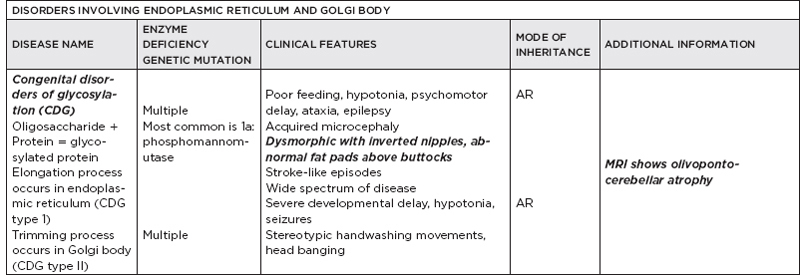
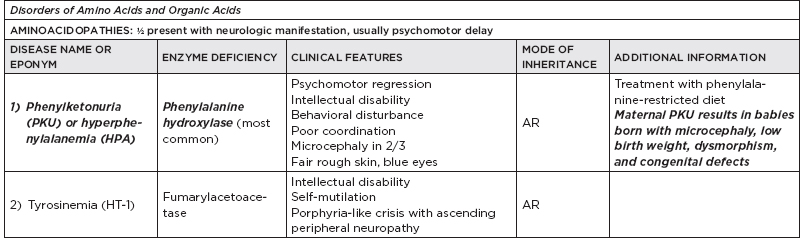
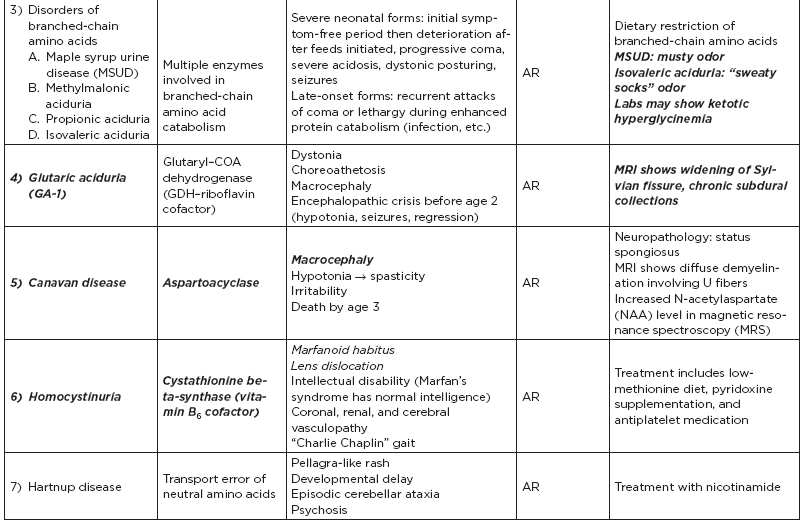
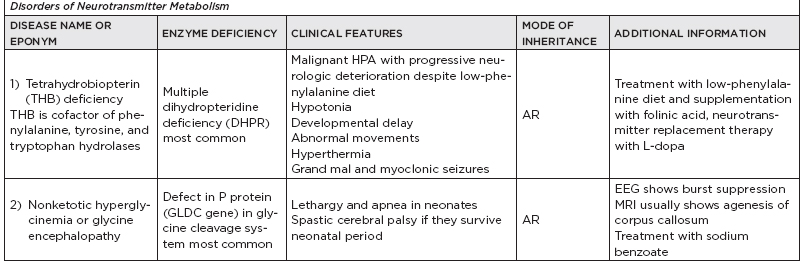
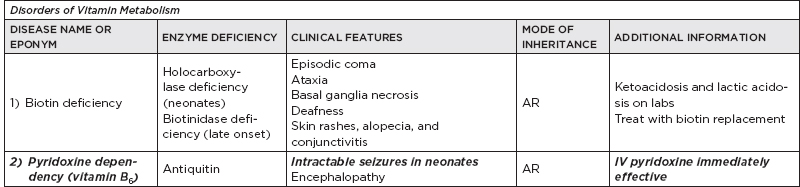

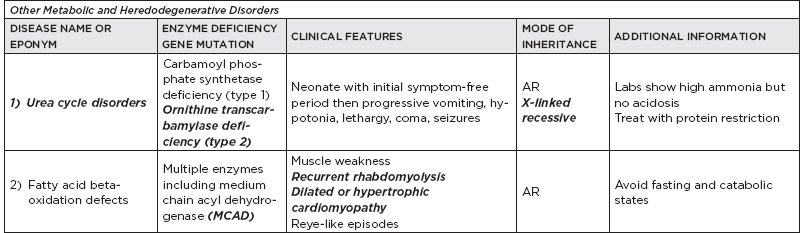
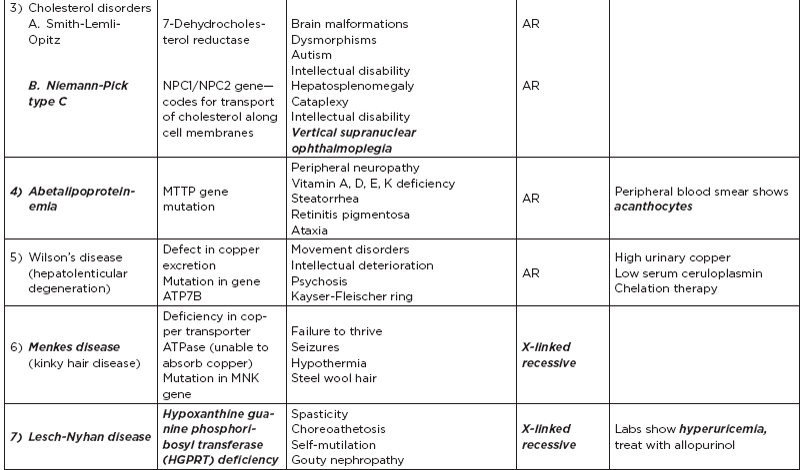


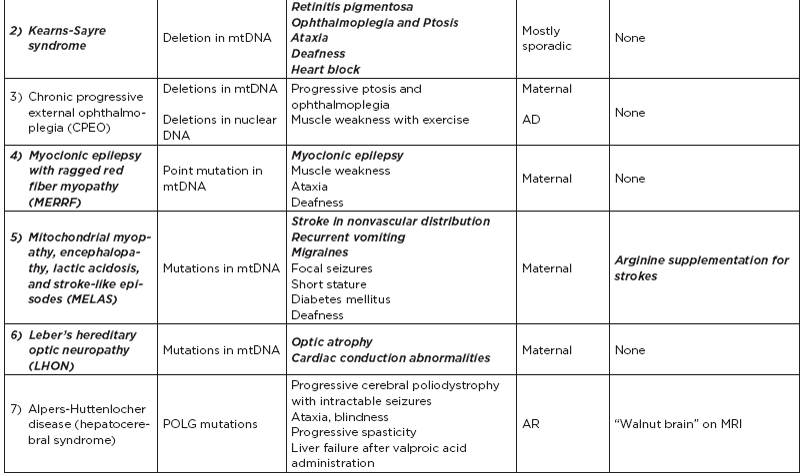
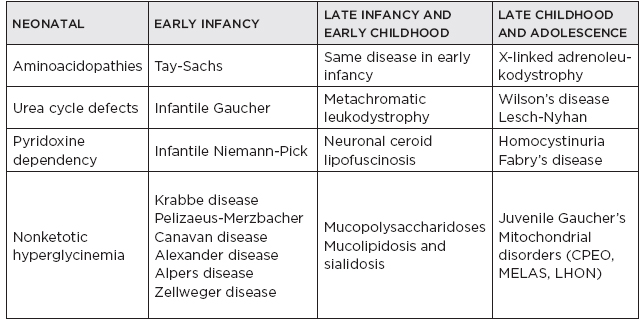
C. Metabolic diseases by age of presentation
IV. Neuromuscular Disorders
A. Neuropathies
1. Hereditary motor and sensory neuropathies (HMSN) type 1: Charcot-Marie-Tooth, peroneal muscular atrophy; all types generally have insidious clinical onset and slow progression from adolescence; rarely, they can present in infancy; pes cavus and hammer toes often cause initial complaints; segmental demyelination and remyelination occur, resulting in distal muscle atrophy and weakness and tremor and ataxia in some (39%); autosomal dominant; multiple genetic subtypes; in older patients, nerve biopsy shows a hypertrophic onion bulb appearance; may have elevated CSF protein; life expectancy is normal; duplication of PMP22.
2. HMSN type 2a: autosomal dominant; map to chromosome 1; axonal neuropathy; milder course compared to type 1; HMSN type 2b: childhood onset; autosomal recessive
3. HMSN type 3 (Dejerine Sottas): autosomal recessive; presents at birth; may be a homozygous form due to a sporadic point mutation; hypotonia and slow motor development are common in the first year; sensory ataxia develops; clubfoot and scoliosis are seen; usually with elevated CSF protein.
4. HMSN type 4 (Refsum’s): autosomal recessive deficiency of phytanic acid oxidase affects lipid metabolism; onset is 1st to 3rd decade with cerebellar ataxia, chronic hypertonic neuropathy, and retinitis pigmentosa; other findings: night blindness, deafness, ichthyosis, cardiac myopathy, hepatosplenomegaly, and increased CSF protein; dietary restriction of phytanic acid (avoiding nuts, spinach, and coffee) is beneficial, as phytanic acid is not produced endogenously; infant and adult forms are seen.
5. Hereditary neuropathy with liability to pressure palsies (tomaculous neuropathy): 10% have deletion of PMP-22 protein.
B. Anterior horn cell/muscle disorders
1. Infantile spinal muscular atrophy: three types, all related to chromosome 5; frequency of carriers is 1 in 60; prenatal screening available.
a. Werdnig-Hoffman: infantile form; autosomal recessive; presents at birth with proximal hypotonia and respiratory insufficiency; reduced fetal movement, hypotonia, areflexia, quivering tongue; progressive feeding difficulty and death can occur by age 6 months; muscle biopsy is also diagnostic.
b. Kugelberg-Welander: chronic form; autosomal recessive or sporadic; presents after 3 months with pelvic girdle weakness and runs a variable course; mean survival is 30 years.
c. Third form affects primarily the neck and respiratory muscles; presenting with head droop; survival to age 3 years.
2. Neurogenic arthrogryposis: sporadic disease; affects fetus, causing contractures by the time of birth; electromyography (EMG) is normal but shows a neuropathic process.
3. Fazio-Londe: onset in early childhood; progressive bulbar paralysis, with anterior horn cell involvement
4. Glycogen storage diseases: autosomal recessive
a. Type 2 (Pompe’s): deficient acid maltase activity (1,4 glycosidase) results in glycogen deposition in the anterior horn cells; infantile form presents as floppy infant with congestive heart failure, macroglossia, hepatomegaly; muscle biopsy shows periodic acid–Schiff-positive deposits and vacuolation.
b. Type 3 (Forbes-Cori): debrancher enzyme (1,6 glucosidase) deficiency associated with hypotonia, hypoglycemia, hepato-/cardiomegaly; prognosis is variable; skeletal and cardiac muscles affected.
c. Type 5 (McArdle’s): results from inactive myophosphorylase; childhood and adult forms seen; exercise induces painful cramps; ischemic exercise test shows no lactate production; biopsy shows periodic acid–Schiff-positive subsarcolemmal blebs or crescents.
d. Tarui’s (type 7): phosphofructokinase deficiency results in cramping and fatigue.
![]() NB:
NB:
McArdle’s disease and Tarui’s disease do not produce lactate in the exercise ischemic test.
e. Nonmyopathic types: type 1 (von Gierke; deficient glucose-6-phosphate causes neonatal seizures); type 4 (Anderson’s; deficiency of 1,4 debrancher enzyme results in failure to thrive); type 6 (Hers’; liver phosphorylase deficiency results in growth retardation)
5. Muscular dystrophies
a. Paramyotonia congenita: autosomal dominant; defect on chromosome 17q23.1 affects voltage-gated Na+ channels; causes myotonia on exposure to cold; electrolytes are normal; compare to: myotonia congenita (Thomsen’s disease)—autosomal dominant on chromosome 7; mutation of the chloride channel; seen at birth, muscle hypertrophy (mini-Hercules); EMG: myotonic discharges
b. Duchenne’s muscular dystrophy: the most common dystrophy, affects boys by age 5 years; incidence is 1 in 3,500; 30% mutation rate; localized to Xp21; defects in the gene for dystrophin results in variable amounts of this essential muscle structural protein; weakness, pseudohypertrophy of the calf muscles and tendon shortening are classic; mild MR and cardiac involvement are also present; treatment: prednisone may improve strength and function; creatine phosphokinase (CPK) is elevated; death usually by age 20 years; biopsy: atrophy and hypertrophy, central nuclei, fiber splitting, necrosis, fibrosis, fatty changes, and hyaline fibers; EMG: myopathic units denervation, fibrillation and sharp waves.
c. Becker’s dystrophy: also X-linked; but milder defect, slower progression
d. Limb-girdle dystrophy: autosomal recessive (chromosome 15), autosomal dominant (chromosome 5), severe childhood autosomal recessive muscular dystrophy (chromosome 13); slowly progressive proximal weakness: iliopsoas, quadriceps, hamstrings, deltoids, biceps, triceps; facial and extraocular muscles spared; slightly elevated CPK; EMG: myopathic changes; pathology: fiber size variations; fiber splitting; degeneration/regeneration
e. Fascioscapulohumeral dystrophy: autosomal dominant; on chromosome 4; onset at end of the 1st decade; slowly progressive weakness of facial musculature (Bell’s phenomenon); serratus anterior (winging of the scapula) and biceps; deltoid and forearm muscles preserved (giving Popeye appearance); scapuloperoneal form: on chromosome 5; CPK slightly elevated; EMG and pathology: myopathic changes.
f. Congenital muscular dystrophy: rare; onset at age 2 to 3 years
g. Emery-Dreifuss (humeroperoneal): X-linked recessive (most common) but also has other inheritance patterns; weakness over biceps, triceps, distal leg; contractures early, cardiac conduction block
h. Oculopharyngeal dystrophy: common in French Canadians or Spanish Americans; autosomal dominant; onset in 5th decade, slowly progressive; ptosis first, pharyngeal weakness later; CPK slightly elevated; pathology: myopathic changes, rimmed vacuoles, and intranuclear tubulofilamentous inclusions
i. Myotonic dystrophy: autosomal dominant; cytosine-thymine-guanine (CTG) triplet repeat (>50 copies on chromosome 19q); related to defective protein kinase and membrane instability; it is a multisystem disease that usually presents in adults (20–40 years old [y/o]), not in children; results in facial weakness (ptosis, fish mouth), hatchet face, some MR, posterior capsule cataracts, cardiac disease, diabetes, testicular or ovarian atrophy; congenital form is severe at birth but improves in 4 to 6 weeks; EMG: spontaneous bursts of high frequency amplitude discharges; pathology: type 1 fiber hypertrophy and ring fibers; congenital myotonic dystrophy in children of mothers with myotonic dystrophy.
6. Myopathies
a. Nemaline: autosomal recessive on chromosome 1; occasionally autosomal dominant; non-progressive; also high-arched palate, small jaw and thin face; Marfanoid features, cardiomyopathy; CPK is normal; type 1 fiber predominance with Z-line rods.
b. Central core: autosomal recessive on chromosome 19; floppy baby, motor delay, spine abnormalities, proximal, nonprogressive; CPK is normal; type 1 fibers have central pallor; on electron microscopy: core lacks mitochondria.
c. Myotubular (centronuclear): X-linked recessive; age of onset is 5 to 30 years; involvement of ocular, facial, and distal muscle; variable progression; CPK is normal or mildly increased; biopsy: central nuclei with halos and type 1 fiber atrophy.
d. Dermatomyositis: affects females more than males; skin lesions: diffuse erythema, maculopapular eruption, heliotrope rash, eczematoid dermatitis of extensor surface joints; carcinoma in 15% (affects more adults than children); lab: CPK high, aldolase high, IgG and IgA levels may be elevated; myoglobinuria; inflammatory muscle changes; sometimes tissue calcification.
e. Hypokalemic periodic paralysis: may be autosomal dominant or associated with thyrotoxicosis; age between 10 and 20 years; attacks are frequent and usually severe, lasting for hours to days; trigger: rest, cold, stress; low serum K+; calcium channelopathy; treatment: acetazolamide, K+ replacement.
f. Hyperkalemic periodic paralysis: autosomal dominant; age between 10 and 20 years; attacks are frequent with moderate severity lasting minutes to hours; triggers: rest, cold, hunger; high serum K+, occasional myotonia, Na+ channelopathy; treatment: acetazolamide, low potassium.
C. Neuromuscular junction disorders
1. Neonatal (transient) myasthenia: transient disorder seen in 15% of infants born to mothers with myasthenia gravis; due to placental transfer of acetylcholine receptor (AChR) antibodies; symptoms: intrauterine hypotonia; may be born with arthrogryposis; usually evident within 24 hours of life, lasting for 18 days (range, 5 days to 2 months); may need exchange transfusion and/or neostigmine, 0.1 mg intramuscularly before feeding.
2. Congenital myasthenia: heterogeneous disorder due to genetic defects in the presynaptic (mostly autosomal recessive) and postsynaptic (mostly autosomal recessive, some autosomal dominant [slow channel syndrome]) neuromuscular junction; not associated with antibodies to AChR; symptoms: usually begin in the neonatal period, ocular, bulbar, respiratory weakness, worse with crying or activity; ptosis, ophthalmoplegia, or ophthalmoparesis; diagnosis: positive family history in some, Tensilon® (edrophonium chloride) test negative in most, AChR is negative, EMG: decremental response and increased jitter.




