Pharmacology
Neurotransmitters and Receptors
I. Acetylcholine
Synthesis: occurs in the nerve terminal
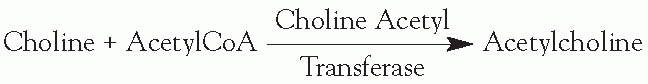
This is the rate-limiting step in production of acetylcholine (ACh).
The supply of choline is the rate-limiting factor in production.
Release: Voltage-dependent calcium (Ca) channels open with depolarization, causing an influx of Ca.
This results in fusion of the synaptic vesicles and release of neurotransmitter.
Degradation: occurs in the synaptic cleft

Reuptake: Choline is recycled by the presynaptic terminal.
Receptors
Muscarinic (M) receptors (in brain, muscarinic > nicotinic )
All subtypes linked to G-proteins
M 1,3,5 activate phosphytidyl inositide hydroxylase
M 2,4 inhibit adenyl cyclase
Muscarinic agonists (cholinergic, parasympathomimetic)
Bethanechol—bladder (not degraded by esterase), carbechol—gut
Pilocarpine—eye
Methacholine
Muscarinic antagonists (anticholinergic)
Atropine, scopolamine, tricyclic antidepressants
Pupils dilate, tachycardia, reduced secretions, decreased sweating, increased intraocular pressure
May help control tremor and rigidity in Parkinson disease
Nicotinic receptors (N1 through N4)
Central nervous system (CNS) sources
Nucleus basalis of Meynert
Diagonal band of Broca
Caudate
Locations
Nicotinic and muscarinic
All preganglionic synapses (sympathetic and parasympathetic)
CNS (M>N)
Muscarinic
All postganglionic parasympathetic
Postganglionic sympathetic at sweat glands. (The rest of the postganglionic sympathetic synapses use epinephrine and/or norepinephrine.)
Nicotinic
NMJ
Adrenal medulla
Disease states
Acetylcholine deficiency
NMJ release blockade (presynaptic disorders)
Botulinum toxin—blocks presynaptic vesicle mobility
Lambert-Eaton syndrome—blocks presynaptic calcium channels
Tick paralysis
Sea snake toxin
NMJ receptor blockade (postsynaptic disorders)
Myasthenia gravis—ACh receptor antibodies
Depolarizing blockade—succinylcholine
Nondepolarizing blockade—curare, procainamide, aminoglycosides
α-Bungarotoxin—irreversible ACh receptor blockade
Alzheimer disease
Degradation of ACh nuclei in nucleus basalis
Basal forebrain atrophy
Acetylcholine excess
Anticholinesterases (acetylcholinesterase inhibitors)
Prevent breakdown of ACh at the synaptic cleft and increase amount of ACh available in the cleft. Some examples:
Pyridostigmine (Mestinon)—used in myasthenia gravis
Physostigmine
Edrophonium (Tensilon)
Tacrine, donepezil—used in Alzheimer disease
Organophosphates, diisopropyl fluorophosphate, sarin, soman—irreversible action at the receptors
Black widow spider venom
Causes explosive release of ACh
β-Bungarotoxin
Promotes release of ACh
II. Norepinephrine (NE)
Synthesis
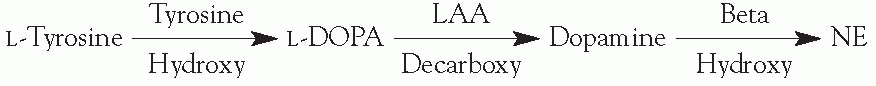
Rate-limiting step is mediated by tyrosine hydroxylase.
NE is a feedback inhibitor of tyrosine hydroxylase.
L-aromatic amino acid (LAA) decarboxylase requires a vitamin B6 cofactor.
Dopamine β-hydroxylase is a copper-containing enzyme and requires oxygen and vitamin C as cofactors.
NE is converted to epinephrine by phenylethanolamine-N-methyl transferase in the adrenal medulla only.
Storage
Dopamine (DA) and NE are transported into the vesicles by a magnesium (Mg) and ATP-dependent process.
Transport into vesicles is inhibited by reserpine and tetrabenazine.
Oxidation of DA to NE occurs in the vesicles.
NE is displaced from the vesicles by amphetamines and ephedrine.
Release
Vesicles are released after depolarization results in calcium influx.
Catecholamines in the synaptic cleft then inhibit further vesicle release.
Amphetamines increase release.
Metabolism
Metabolism of catecholamines occurs more slowly than does ACh metabolism. Catechol-O-methyl transferase (COMT) and monoamine oxidase (MAO) are the two major enzymes in catecholamine metabolism.
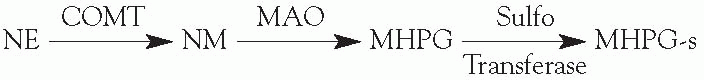
MAO
Found on the outer surface of presynaptic mitochondria and on the postsynaptic cell membrane. MAOb is found primarily in the CNS.
MAOa is blocked by clorgyline and pargyline.
MAOb is blocked by selegiline and pargyline.
COMT
Found only on the postsynaptic cell membrane
COMT is blocked by tropolone.
Reuptake
Reuptake is the primary mode of NE termination.
Reuptake is mediated by sodium (Na)/ATP channel.
Reuptake is inhibited by cocaine, nortriptyline, amitriptyline, imipramine, and desipramine.
Receptors
All receptors work via G-proteins.
α1-Receptor is most sensitive to epinephrine and blocked by prazosin and clonidine. It is postsynaptic
α2-Receptor inhibits adenyl cyclase and is inhibited by yohimbine and clonidine. It is presynaptic.
Phentolamine and phenoxybenzamine—block both α1-receptors and α2-receptors
Labetalol—blocks both alpha and beta receptors
Location
Locus ceruleus
Hypothalamus
Reticular activating system
III. Dopamine
Synthesis
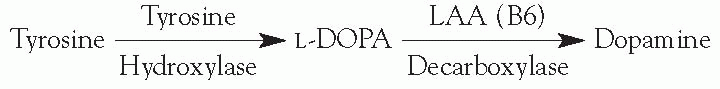
Tyrosine hydroxylase (TH) mediates the rate-limiting step.
LAA decarboxylase is a vitamin-B6-dependent enzyme.
DA is a feedback inhibitor of TH and of the release of vesicles.
Release: action potentials cause calcium influx, which results in the fusion of vesicles.
Metabolism: after reuptake, presynaptic intraneuronal MAO converts DA to DOPAC (dihydroxyphenylacetic acid). Extraneuronal postsynaptic COMT and MAO convert DA to homovanillic acid (HVA).
Reuptake: presynaptic terminals contain high-affinity DA transporters.
Receptors
D1,5
Postsynaptic linked to G-protein
Excitatory
Stimulates cAMP (cyclic adenosine monophosphate)
D2,3,4
Presynaptic—inhibitory (high affinity)
Postsynaptic—inhibitory (low affinity)
DA Pathways
Nigrostriatal—substantia nigra to striatum
Tuberoinfundibular
Mesolimbic—ventral tegmentum to limbic areas/nucleus accumbens
Mesocortical—ventral tegmentum to prefrontal cortex
Medications
Antipsychotics
Dopamine blockers (D2). The antipsychotics block the inhibitory D2-receptors with a resultant excitatory effect. D2-receptor affinity correlates with efficacy.
Amphetamines
Increase release and decrease reuptake of dopamine
MAO inhibitors—antidepressants
Increase DA by decreasing metabolism
Examples: selegiline, pargyline
Cocaine and tricyclic antidepressants
Block reuptake
Reserpine and tetrabenazine
Prevent uptake of DA and NE into vesicles
Model of Parkinson disease (MPTP)
MAO converts MPTP to MPP+ (toxic to dopaminergic cells). This is the basis for using selegiline to treat Parkinson disease.
IV. Serotonin
Synthesis
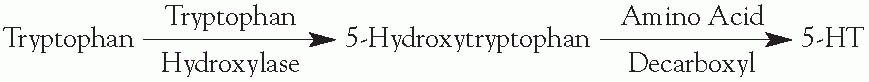
Tryptophan hydroxylase mediates the rate-limiting step.
Increasing the level of L-tryptophan increases the production of 5-hydroxy tryptamine (5-HT).
Amino acid decarboxylase requires a vitamin B6 cofactor.
Storage
5-HT complexes with proteins, divalent cations, and adenosine diphosphate in granules.
Storage is disrupted by reserpine and tetrabenazine.
Release
Amphetamine and fenfluramine cause the release of 5-HT.
Clomipramine and amitriptyline both increase release and block reuptake of 5-HT.
Metabolism

Reuptake: is the primary mechanism of inactivation; is blocked by clomipramine, amitriptyline, sertraline, and fluoxetine.
Receptors
5-HT1a
Linked to G-protein, inhibiting adenyl cyclase
Agonist—buspirone
5-HT1b/d
Linked to G-protein, inhibiting adenyl cyclase
Both act as autoreceptors
Agonist—sumatriptan (5-HT 1d)
5-HT1c
Only type 1 receptor that has an antagonist; really more like a 5-HT2
Linked to G-protein, activating PLC to increase DAG and IP3
Agonist—α-methyl-5-HT, (LSD)
Antagonist—retanserine, pizotofen, clozapine. Note: This is the exception to the no antagonist rule for 5-HT1 receptors.
5-HT2
Linked to a G-protein activating PLC to increase DAG and IP3
Agonist—alpha-methyl-5-HT, (LSD)
Antagonists—ketanserine, pizotifen, clozapine
5-HT3
Agonists—2-α-5-HT
Antagonists—metoclopramide, cocaine (weak), ondasetron (potent)
CNS Source: midline raphe nuclei
V. Glutamate
Excitatory amino acid neurotransmitter
Receptors
N-methyle-D-aspartate (NMDA)
Activates mainly calcium channels
Five binding sites alter channel opening
Glutamate—increases
Glycine—increases
Polyamine—increases
Mg—decreases flow
Zinc—decreases flow
Glutamate and glycine are required for channel opening.
Voltage-dependent blockers—phencyclidine, ketamine, Mg, MK-801
Voltage-independent blockers—zinc
Found in hippocampal pyramidal cells
Excitotoxicity theory—Normally the NMDA receptor is blocked by Mg. With enough glutamate binding, the Mg-induced blockade can be overcome and calcium is allowed to enter the cell. This activates intracellular biochemical processes, which can lead to cell destruction.
2-(Aminomethyl)phenylacetic acid (AMPA)
Activate mainly sodium channels
Major source of CNS fast excitatory postsynaptic potentials (EPSPs)
Receptor affinity—AMPA > glutamate > kainate
GluR3 receptor implicated in Rasmussen encephalitis
Kainate
Receptor affinity—kainate > Glutamate > AMPA
APCD
L-AP4—G-protein coupled formation of AMP
Inactivation
Reuptake is primary mode of termination.
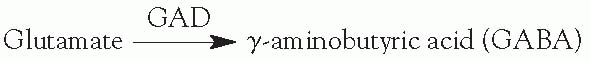
Glutamic acid decarboxylase (GAD) is a vitamin-B6-requiring enzyme.
B6-responsive seizures—decreased B6 can lead to increased glutamate and decreased
GABA, and in theory, can lead to increased seizures.
Administration of vitamin B6 helps some children with seizures.
VI. GABA
Inhibitory amino acid neurotransmitter
Receptors
GABA-a—fast inhibitory postsynaptic potentials (IPSPs). Increases chloride conductance.
Has five binding sites:
Benzodiazepines increase the frequency of chloride channel opening.
Barbiturates prolong the duration of chloride channel opening.
Steroid site
Picrotoxin site—blocker (model for epilepsy)
GABA
Locations—cortex, hippocampus, basal ganglia
GABA-b—slow IPSPs. Increases potassium conductance.
Coupled to G-proteins, which use adenyl cyclase as a second messenger
Agonist—baclofen
Antagonist—phaclofen
Location—cerebellum, spinal cord
Inactivation
Reuptake
Enzymatic degradation:
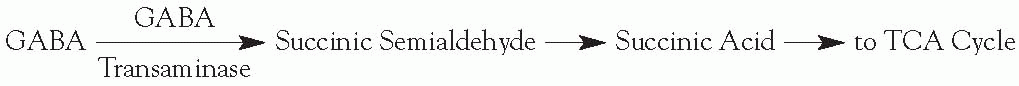
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree



