An axial T1 and FLAIR MRI brain scan shows numerous white matter lesions involving cerebral hemispheres ovoid in shape and periventricular in location, which is consistent with multiple sclerosis. Many white matter lesions demonstrated T1 hypointensity consistent with neuronal loss (”T1 black holes”). Mild generalized cerebral volume loss was noted. The pons and left middle cerebellar peduncle were also involved (not shown).
Three-step assessment
1 Classical clinical features of MS: hemiataxia with resolution, dementia
2 Neurological examination: dementia with corticospinal tract impairment (extensor plantar reflex)
3 Investigations: MRI brain consistent with MS; CSF consistent with MS
Diagnosis: Dementia due to multiple sclerosis.
Tip: Cognitive impairment due to multiple sclerosis can occur without significant motor or sensory impairment, even over many years. Cognitive impairment may be severe enough to warrant a diagnosis of significant dementia. Patients that smoke cigarettes appear to be overrepresented in the population of MS patients with significant dementia.
Case 36: A woman with MS and severe memory loss
A 53-year-old woman had transient true spinning vertigo lasting one week six years prior to evaluation. The following year she developed right upper extremity numbness and significant gait ataxia, again with resolution. Two years following that, she had insidiously progressive memory impairment. She was unable to manage her own finances for the prior four years, and she could not prepare meals any longer as she would forget the ingredients. She would leave tap water running and forget to turn it off. She could not reliably dress herself independently and she discontinued driving. She was poor at calculations, could not write, or focus her attention to play cards. She had a history of mood disorder with depression and anxiety, but this was stable and had not worsened.
On neurological examination, she scored 14 out of 38 on a Kokmen short test of mental status, displaying abnormalities on orientation, attention, registration, calculation and delayed recall. On language testing, she had impaired comprehension, mild difficulty with naming and moderate difficulty with repetition and writing. She had evidence of upper extremity ideomotor apraxia and palmar grasp response bilaterally with some pseudobulbar affect. A motor examination was normal but plantar responses were extensor bilaterally. Her walking revealed gait initiation failure (gait “apraxia”).
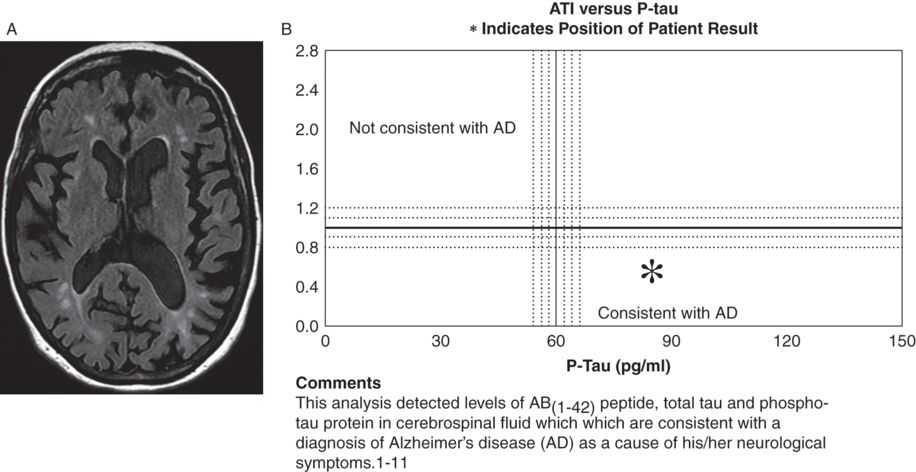
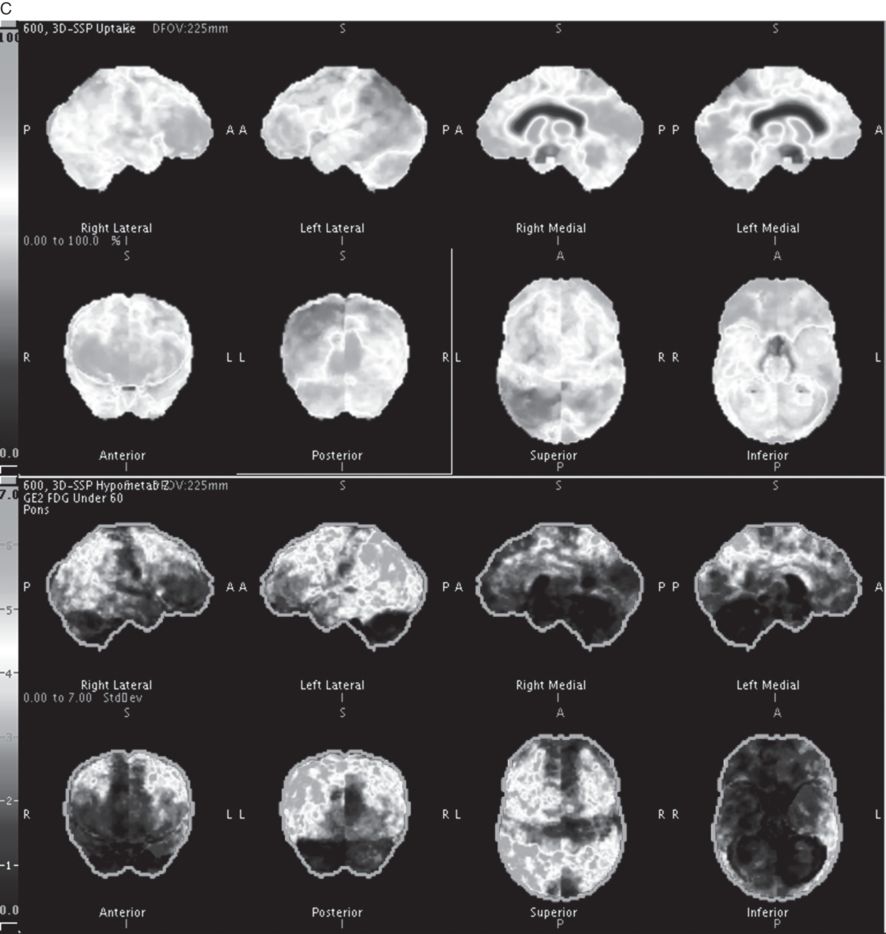
Brain and spinal cord (not shown) MRI scans were highly consistent with multiple sclerosis (Figure 6.2 A). A CSF examination showed elevated oligoclonal bands and revealed tau and amyloid-b1–42 metabolites and amyloid-b1–42/tau index changes, reflecting a high sensitivity 85–94 percent and specificity 83–89 percent for Alzheimer’s disease (Figure 6.2 B). A brain 18 F-fluorodeoxyglucose-PET (FDG-PET) revealed bilateral (left greater than right) fronto-temporal-parietal and posterior cingulate hypometabolism, highly suggestive of Alzheimer’s disease (Figure 6.2 C).
A) Axial T1 and FLAIR MRI brain scans showed periventricular and subcortical scattered T2 hyperintensities oriented perpendicularly to the lateral ventricles with moderate cerebral atrophy with bilateral frontoparietal predominance, slightly more pronounced on the left. A cervical spine MRI (not shown) demonstrated multiple discontinuous foci of T2 hyperintensity from the C2 level to C5 involving the posterior and/or lateral aspects of the spinal cord. B) CSF revealed tau and amyloid-b1–42 metabolites and amyloid-b1–42/tau index changes reflecting a high probability of Alzheimer’s disease. C) Brain 18 F-fluorodeoxyglucose-PET (FDG-PET) showed bilateral (left greater than right) fronto-temporal-parietal and posterior cingulate hypometabolism, highly suggestive of Alzheimer’s disease.
Three-step assessment
1 Classical clinical features of MS: vertigo, ataxia, hemisensory symptoms with resolution, progressive severe dementia
2 Neurological examination: very severe dementia with corticospinal tract impairment
Diagnosis: Multiple sclerosis with Alzheimer’s disease.
Tip: Cortical symptoms and more severe signs of dementia may herald an alternative accompanying dementing disorder in addition to a history of multiple sclerosis. Novel new biomarkers for Alzheimer’s disease including brain FDG-PET and CSF tau and Abeta metabolites can diagnose this condition in life. Occasional cases of MS complicated by Alzheimer’s disease have been confirmed at autopsy. Confirmation of this unusual presentation may provide opportunities to treat Alzheimer’s disease confidently with acetylcholinesterase inhibitors that have proven but modest benefits.
Case 37: A middle-aged man with progressive, treatment-resistant hemiparesis
A 52-year-old man presented with progressive left facial weakness, followed over the next few weeks and months by slowly progressive left upper and lower extremity weakness with dysarthria. He also had a significant personality change; specifically, he was previously felt to be very driven and ambitious and was now far more “easygoing.” He had no other symptoms of MS attacks. There was no family history of MS. Intravenous corticosteroids were given without any improvement. He initiated treatment with interferon beta-1A intramuscular injections weekly along with monthly prednisolone infusions for a suspected diagnosis of MS. He started to use a cane initially and before long required a wheelchair.
On neurological examination he was inattentive and mildly inappropriate but was found to have no other impairment on mental status examination. He had no visuospatial neglect and his visual fields were full, and he had no papilledema. He had saccadic breakdown of smooth pursuit eye movements, and spastic dysarthria. A motor examination revealed left upper motor neuron-type facial weakness with severe spastic left hemiparesis. Right motor power was normal. Plantar responses were extensor on the left and flexor on the right. Cortical-type and other sensory exams were normal. He was wheelchair bound.
A brain MRI showed greater T2 signal abnormality in the right than left hemisphere white matter with an area of abnormal T2 signal within the pons (Figure 6.3). There was evidence of mass effect as well as subtle gadolinium enhancement. Cervical and thoracic spinal cord MRIs were normal. A CSF examination was normal without oligoclonal bands or elevations in IgG index and a cytology was negative for malignancy. A CSF was negative for Lyme disease, Whipple’s agent, syphilis, angiotensin converting enzyme (ACE), tuberculosis, West Nile virus, herpes simplex virus and John Cunningham (JC) virus polymerase chain reaction (PCR). Fungi and bacterial cultures were all negative. A brain biopsy confirmed pathological evidence of gliomatosis cerebri.
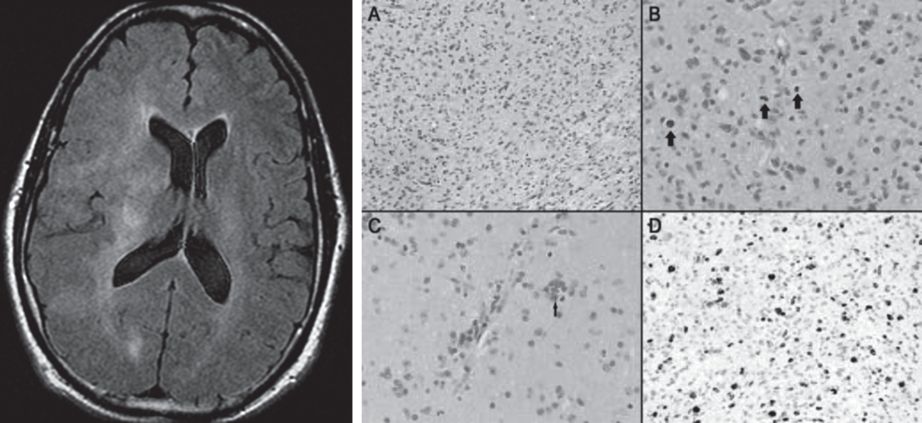
A) Axial FLAIR MRI shows a bilateral, asymmetric diffuse white matter process that extends from the pons through the midbrain (not shown) into the deep white matter of both cerebral hemispheres. There is mass effect with effacement of several sulci and shift of midline structures to the left. The imaging findings are most consistent with a diffusely infiltrative process such as a glioma. B) Hematoxylin eosin stains show elongated, pleomorphic neoplastic cells diffusely infiltrating cerebral parenchyma with evidence of perineuronal satellites, perivascular neoplastic aggregates and numerous mitotic figures.
Three-step assessment
1 Classical clinical features of MS: progressive hemiparesis, but treatment-resistant with personality change
2 Neurological examination: upper motor neuron hemiparesis with inattention and mildly inappropriate behavior
3 Investigations: MRI brain not consistent with MS; MRI spinal cord not consistent with MS; CSF not consistent with MS; brain biopsy confirmatory of primary brain neoplasm
Diagnosis: Gliomatosis cerebri.
Tip: The patient progressed to death 14 months later. The progressive nature of the patient’s worsening that was treatment-resistant along with atypical MRI features, a normal CSF and a normal MRI scan of the spine indicated an alternative diagnosis to that of multiple sclerosis. Gliomatosis cerebri is a diffusely infiltrating astrocytoma that affects multiple lobes. Mass effect and gadolinium enhancement may not be seen, particularly early in the clinical course. Median survival has been estimated at between 6 and 39 months.
Case 38: A man with progressive white matter disease and an “alien” limb
A 41-year-old man presented with progressive left upper and lower stiffness. He exhibited left upper extremity limb apraxia and “alien limb” phenomenon, which manifested as his hand involuntarily grabbing onto his shirt repetitively. He developed bilateral lower extremity weakness as well as a spastic dysarthria. He had no family history of any neurological diseases including MS or leukoencephalopathy. Serological testing was negative for inherited, acquired and other degenerative leukoencephalopathies known at the time.
On neurological examination he had mild impairment on a Kokmen short test of mental status, scoring 34/38 and missing elements on orientation, construction and delayed recall. Cranial nerves were normal, including color vision. He had an upper motor neuron spastic dysarthria, with a brisk jaw jerk. He had a moderate left greater than right hemiparesis, with normal sensation walking with a left hemiparetic gait. He had a grasp reflex and apraxia of the left upper extremity.
Serial brain MRIs showed progressive, asymmetrical confluent white matter abnormalities without gadolinium enhancement (Figure 6.4 A). Cervical and thoracic spinal cord MRI scans were normal. A CSF examination was normal apart from a moderate increase in neuron-specific enolase (NSE) without elevations in either oligoclonal bands or IgG index. A brain biopsy showed axonal spheroids appearing as pale eosinophilic globules on hematoxylin and eosin staining (Figure 6.4 B). An electron microscopy of the brain pathology demonstrated axonal spheroids within the white matter (Figure 6.4 C).
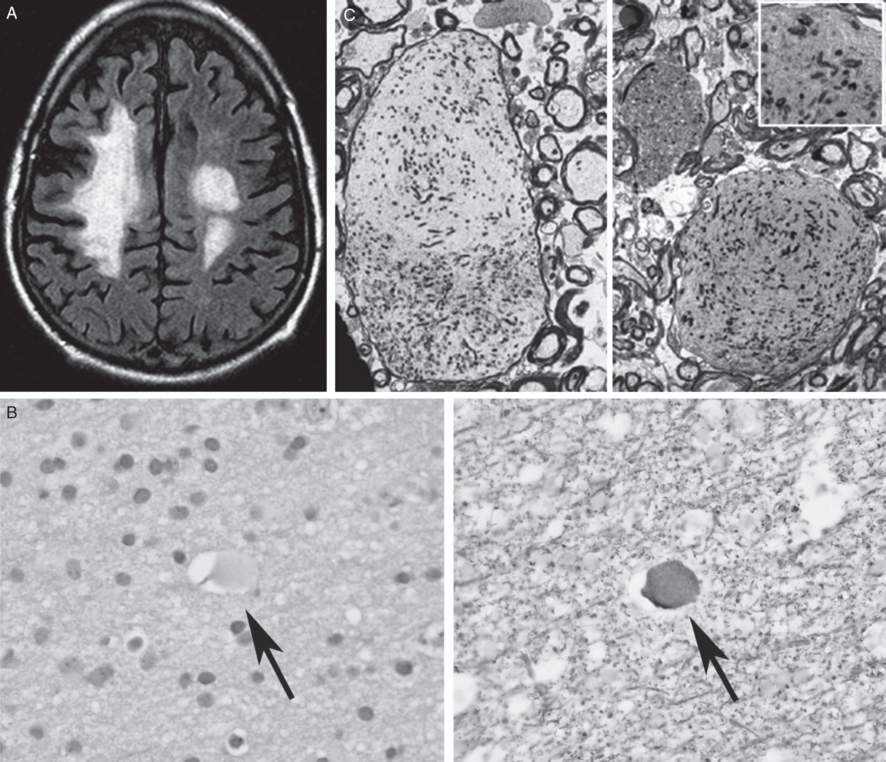
A) Axial FLAIR MRI brain showing moderate to marked confluent non-enhancing T2 foci in the right greater than left cerebral hemispheric white matter (from BM Keegan et al., 2008 with permission). B) Brain biopsy showing axonal spheroids appearing as pale eosinophilic globules on hematoxylin and eosin staining. C) Electron microscopy of the brain pathology demonstrated axonal spheroids within the white matter (from BM Keegan et al., 2008, with permission).
Three-step assessment
1 Classical clinical features of MS: progressive upper motor neuron quadriparesis
2 Neurological examination: asymmetric upper motor neuron quadriparesis with alien limb phenomenon and signs of frontal lobe dysfunction
3 Investigations: MRI brain not consistent with MS; MRI spinal cord not consistent with MS; CSF not consistent with MS; brain biopsy confirmatory of sporadic adult onset leukoencephalopathy with neuroaxonal spheroids
Diagnosis: Sporadic adult-onset leukoencephalopathy with neuroaxonal spheroids mimicking multiple sclerosis.
Tip: Important considerations in this case are the progressive nature of the impairment, confluent white matter abnormalities associated with significant atrophy, the lack of gadolinium-enhancing lesions or focal lesions and the lack of spinal cord MRI findings as well as a lack of abnormalities on CSF examination. Brain MRIs may show focal areas of restricted diffusion, particularly in patients with rapid onset of sporadic adult-onset leukoencephalopathy with neuroaxonal spheroids. Familial cases of neuroaxonal spheroids with leukoencephalopathy have been described as well. The genetic mutation has been found to be in the colony-stimulating factor 1 receptor (CSF1R) gene, causing hereditary diffuse leukoencephalopathy with spheroids. Whether this genetic mutation is responsible for sporadic cases currently remains uncertain.
Case 39: A man with an abnormal brain MRI and a persistently enhancing cervical spine MRI lesion for possible MS
A 46-year-old gentleman recalled awakening one morning with right arm weakness and numbness. Within about three days, however, the symptoms resolved and these areas returned to normal power and sensation. He would sometimes “lose his train of thought” but had no major cognitive impairment. He had no prior symptoms of optic neuritis, diplopia, dysarthria or dysphagia. He did not have a Lhermitte symptom nor bowel or bladder incontinence, but suffered from urinary hesitancy and incomplete bladder emptying which was attributed to benign prostatic hyperplasia. He had no reported family history of multiple sclerosis, dementia or any other neurological disease. A possible diagnosis of multiple sclerosis was raised and he initiated interferon beta 1-a intramuscular injections once weekly but had to discontinue after getting flu-like symptoms and fever one to three days following the injections.
On neurological examination, he scored 35/38 on a Kokmen short test of mental status. His visual fields were full. Pupils were symmetrical and normal. Extraocular movements were full and normal. Speech was clear. A motor exam was found to be normal and his reflexes were brisk, but his plantar responses were flexor bilaterally. In addition, a sensory exam and his gait and coordination were normal.
A brain MRI showed multiple areas of abnormal T2 signal throughout without enhancement, including signal abnormality in the tips of the temporal lobes bilaterally (Figure 6.5). A cervical spine MRI showed an enhancing lesion at C3-C4 within the cord (Figure 6.6). Repeated cervical spine MRIs continued to show the same enhancing intramedullary lesion at the C3-C4 level that was unchanged in appearance with no associated blood products. An incidental occult vascular malformation such as capillary telangiectasia was therefore suspected. A CSF examination was normal without elevations in oligoclonal bands or IgG index. Visual evoked potentials were normal. A skin biopsy studied by electron microscopy was performed (Figure 6.7). Special attention to dermal vessels demonstrated disorganization of the walls of the vascular smooth muscle cells. The basal lamina was thickened and distorted by irregular deposits of granular osmiophilic material (GOM), all of which features were characteristic of CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy). Genetic testing confirmed a DNA sequence alteration and in the NOTCH3 gene diagnostic of CADASIL.
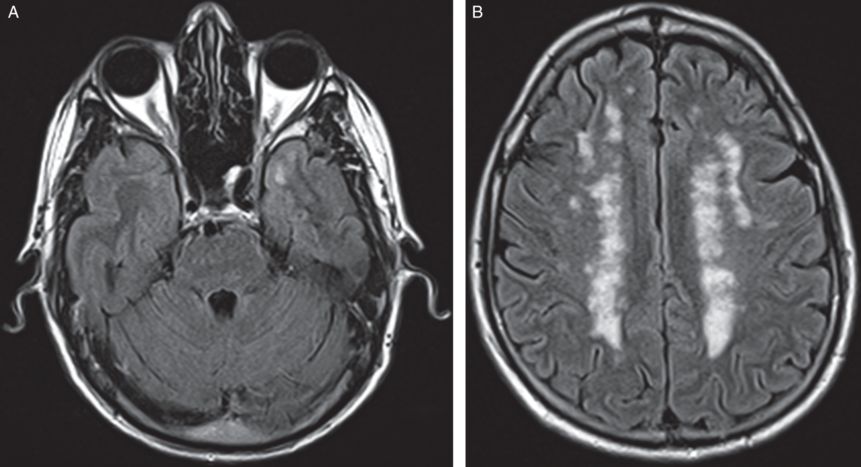
Axial FLAIR MRI shows T2 signal abnormality in the tip of the anterior left temporal lobe. Multiple non-enhancing periventricular and subcortical T2 lesions are seen throughout both cerebral hemispheres.
Related posts:
 Challenges in diagnosing spinal cord disease
Challenges in diagnosing spinal cord disease
 Pitfalls in diagnosing demyelinating cerebellar disease
Pitfalls in diagnosing demyelinating cerebellar disease
 Pitfalls in recognizing uncommon MS clinical presentations
Pitfalls in recognizing uncommon MS clinical presentations
 Challenges in the therapeutic management of MS
Challenges in the therapeutic management of MS
 Challenges in diagnosing demyelinating ocular disease
Challenges in diagnosing demyelinating ocular disease
 Pitfalls in identifying the classical clinical features of MS
Pitfalls in identifying the classical clinical features of MS
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


