Chapter 9 Polyneuropathies
Introduction
Polyneuropathies are generalized disorders of the peripheral nerves and are a common neurological problem. The cause, pathology and presentation of this group of diseases are wide ranging and variable (England & Asbury, 2004). Some neuropathies have an acute onset and progression, whereas others have a more chronic, slower progressing picture. This chapter outlines the most common acute and chronic polyneuropathies, with examples given of disorders according to the structures involved in the pathological process. Evidence and best practice for rehabilitation interventions will be illustrated with a case history.
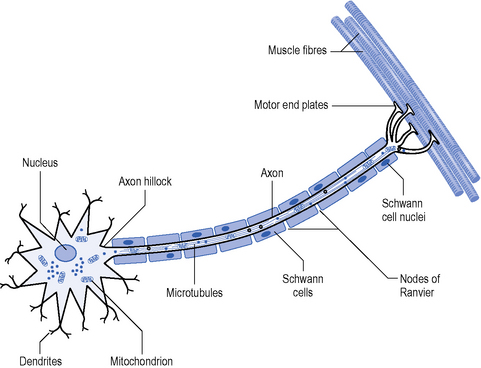
Polyneuropathies are most commonly mixed, affecting both motor and sensory neurones, but some can have a predilection for one modality. The key pathology is an impairment of transmission of nerve action potentials due to disruption of either the axon or the myelin sheath (Figure 9.1). Myelin and axonal damage can occur for a variety of reasons, such as infection, drug or environmental toxicity, metabolic and auto-immune disorders, malignancy and genetic factors (Hughes, 2008).
Background physiology
To understand the pathology of nerve disease, a basic understanding of nerve physiology helps to explain the deficits and symptoms experienced by people with neuropathy (Figure 9.1) (England & Asbury, 2004). The health of the neuron from the proximal to the distal end is maintained by axonal transport. Transport of mitochondrion is important to meet the energy requirements of nerve conduction at the periphery. In fast conducting myelinated neurons, propagation of the action potential relies on the interaction between the nerve axon and myelin. In motor neurons, a clustering of voltage-gated sodium channels occurs at the axon hillock where the initial depolarization occurs. In sensory neurons depolarization occurs through stimulation of the sensory end organ. Clusters of channels also exist at the nodes of Ranvier in myelinated fibres. Action potentials are generated at the nodes by opening of the voltage-gated sodium (Na) channels leading to an influx of Na+ ions which depolarizes the membrane. Closure of the Na+ channels terminates the action potential by restoring the resting membrane potential (Vucic et al., 2009). This process is called saltatory conduction and allows the current to spread further and quicker between nodes so speeding up the action potential.
A commonly used diagnostic tool for investigating polyneuropathy is electrophysiological testing (England & Asbury, 2004). A current is externally applied to a nerve and the speed of conduction, or conduction velocity, can be calculated. This indicates the presence of demyelination as slowing of conduction is seen where saltatory conduction is impaired by myelin degeneration (<38 m/s). Electrical studies also allow the measurement of the amplitude of the motor response, the compound muscle action potential (CMAP), or the sensory nerve action potential (SNAP). A reduction in the amplitude of the CMAP or SNAP indicates a loss of axons. These studies help to distinguish between different types of neuropathy but also help to understand how the structures involved in the pathology contribute to the presentation.
Demyelinating polyneuropathies
Demyelinating neuropathies occur due to degeneration or destruction of myelin. Myelinated nerves tend to be larger and faster conducting, e.g. motor neurons and 1a sensory afferents, and a predilection for large fibre dysfunction is seen in demyelinating neuropathies (England & Asbury, 2004). It is worth noting, however, that prolonged periods of demyelination can lead to axonal degeneration, though the mechanism is not fully understood. Demyelination could cause an alteration of axonal ion channels and/or increased energy requirements that lead to disturbances in axonal transport and subsequent axonal degeneration. There are also reports of down regulation of neurotrophic factors in Schwann cells where structural changes in myelin are observed (Nicholson, 2006).
This section outlines the most common acute and chronic demyelinating polyneuropathies.
Acute demyelinating polyneuropathy
An acute polyneuropathy is described as one that reaches its highest point of severity, or nadir, in less than 4 weeks (Hughes, 2008). The most common type is Guillain Barré syndrome (GBS) or acute inflammatory polyradiculoneuropathy where there is a rapidly progressive paralysis, sensory impairment and areflexia (England & Asbury, 2004). The European incidence is 1.9 cases per 100 000, so it is a relatively rare condition (Vucic et al., 2009). GBS can affect any age group but is more prevalent in men and in older people, where the outcome after the acute period can be much worse (Vucic et,2009).
The cause is not clear but an infection precedes the onset of GBS in about two-thirds of cases (Hughes, 2008; Vucic et al., 2009). GBS is an autoimmune disorder where an immune response is directed towards unknown antigens triggered by the earlier infection. This immune response leads to an inflammatory process and destruction of the myelin sheath in the larger diameter motor and sensory neurons.
Presentation of Guillain Barré syndrome
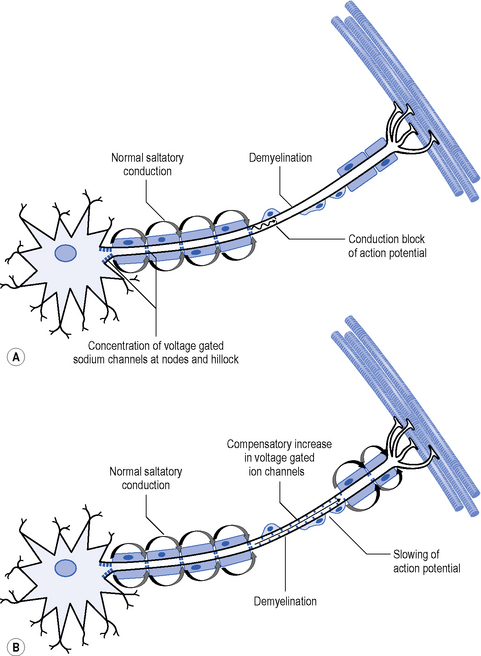
The primary pathology in GBS is one of acute inflammatory demyelination which results in disruption of saltatory conduction leading to a slowing or block of nerve conduction (Figure 9.2). There is also a suggestion that the voltage-gated sodium channels are blocked by antibodies, also impacting on neuron function (Vucic et al., 2009).
GBS presents with proximal or distal weakness and sensory loss that either ascends or descends from onset. Symptoms can impact on functional abilities such as walking, standing or even sitting in severe cases. Facial and bulbar musculature is commonly involved in about 50% of cases (Vucic et al., 2009). In severe cases where there is extensive trunk weakness and bulbar dysfunction, respiratory function and vital capacity (below 20 mL/kg) can be compromised with up to one-third of cases needing respiratory support to maintain lung volumes and manage any secondary respiratory infections (England & Asbury, 2004; Vucic et al., 2009). Tracheostomy is commonly performed and associated with more severe symptoms and extended critical care admission (Ali et al., 2006). Autonomic involvement is commonplace causing fluctuations in blood pressure and cardiac arrhythmias. This can impact on the rehabilitation process, particularly in the early stages.
Urgent diagnosis is required because of the severity of complications. Blood tests demonstrate elevated immunoglobulin anti-bodies in 25% of cases, elevated protein in the cerebrospinal fluid (CSF) is observed in 80% of people with GBS, though this may not be apparent in the first week (Hughes, 2008). Nerve conduction studies show decreased conduction velocity and a partial conduction block due to the demyelination (Figure 9.2), though this may not be apparent in the very early stages. In addition, a decreased CMAP is observed where there is axonal loss (Vucic et al., 2009).
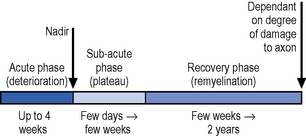
In the early phase the nadir is usually reached in up to 4 weeks with a progressive recovery thereafter (Figure 9.3), though relapses can occur in approximately 16% of cases. Ten to 20% of cases are left with disabling symptoms (Vucic et al., 2009).
Although weakness and sensory loss are the major impairments, persistent fatigue is a common feature in the more chronic stage with 60% reporting severe fatigue after a year. The severity of fatigue, however, does not correlate with disease severity at the nadir, peripheral nerve function or prior infections. It is, however, more prevalent in females and people over 50 years or age (Garssen et al., 2006b; Garssen et al., 2006c). A relationship was found between fatigue and central activation with repeated muscle contraction that may be due to changes in motor unit size and myelination pattern on recovery from GBS (Garssen et al., 2007). A study of Amantadine therapy, a NMDA receptor agonist, was unsuccessful in reducing fatigue symptoms in people with GBS despite some evidence of its efficacy in other neurological disorders (Garssen et al., 2006a).
Pain is frequently reported in 89% of people with GBS that is often severe and may require medical intervention with analgaesics, opioids or anti-depressants such as Gabapentine or Carbomazepine (Hughes, 2008). Pain often originates from the joints and muscles, but there can also be hyperaesthesia (Atkinson et al., 2006).
Management of Guillain Barré syndrome
The initial management of GBS is to address any serious complications, e.g. reduced vital capacity and airway protection through critical care support (see Ch. 15). Drug treatment involves the administration of intravenous immunoglobulins (IVIg) as a first-line intervention with proven efficacy in intervention trials (Hughes, 2008). IVIg can be repeatedly administered, but only if there is a response to the initial intervention (Hughes, 2008). Plasma exchange, or plasmapheresis, is another therapy where the benefits have been established in multiple intervention trials (Vucic et al., 2009). The effect of plasma exchange is to improve disability, with an increased proportion of people making a full recovery and reduction in people requiring mechanical ventilation.
There are no studies of the efficacy of rehabilitation approaches in GBS. At present intervention is based on experience with other neurological conditions though the need for therapy input is recognized (Vucic et al., 2009). Physiotherapy management in the acute stage focuses on respiratory interventions and prevention of secondary complications (Khan, 2004). Respiratory treatments include sputum clearance techniques, maintenance of lung volumes and breathing exercises when the person is able to participate (see Ch. 15). Other interventions advocated are early mobilization, splinting, positioning, stretches to maintain joint range of motion and exercises to increase strength and endurance (Hughes, 2008; Khan, 2004; Meythaler, 1997). Fear, anxiety and sleep deprivation are common problems experienced by people with GBS in the early stages. Good two-way communication between the multidisciplinary team (MDT) and patient and carers is vital to ensure they are informed at every stage; though this may be challenging if a person is intubated, it can be supported by speech and language therapy intervention.
Approximately 40% of all people with GBS will need a period of multidisciplinary inpatient rehabilitation with the aim of maximizing functional recovery and participation (Khan, 2004; Meythaler, 1997). Two studies have found that the more severely affected people tend to access rehabilitation or physiotherapy services (Carroll et al., 2003; Davidson et al., 2009). The impact of therapy input may be important as a recent survey (n>800) of people with GBS found that 90% of people were treated by a physiotherapist, but this fell to 75% of people after discharge from hospital. The survey found that people who did not receive physiotherapy treatment as an outpatient or in the community had greater levels of disability than those who did (Davidson et al., 2009).
Chronic demyelinating polyneuropathy
Chronic demyelinating polyneuropathies can be acquired or inherited and are defined as taking more than 8 weeks to develop (Hughes, 2008). Two of the most common causes are inflammatory and genetic pathologies, and these are described in this section. The efficacy of rehabilitation of the chronic polyneuropathies will be discussed with a case study at the end of the chapter.
Chronic inflammatory demyelinating polyradiculoneuropathy
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is the most common acquired chronic demyelinating neuropathy affecting approximately 3 in 100 000 people (England & Asbury, 2004; Hughes, 2008). It occurs more frequently in adults aged 40 to 60 years with a slightly higher prevalence in men (Westblad et al., 2009).
CIDP has similarities to GBS in that it is an immunologically mediated neuropathy. It presents with multifocal demyelination that predominantly affects spinal nerve roots, proximal nerve trunks and plexuses leading to patchy regions of demyelination with some inflammatory infiltrates (Said, 2006). There is no genetic predisposition to CIDP but a number of studies have suggested an infective episode or vaccination in the weeks prior to onset of the neuropathy. Approximately half of patients have antibodies against myelin-associated glycoprotein and protein levels in the CSF are raised (Said, 2006).
Presentation of chronic inflammatory demyelinating polyradiculoneuropathy
The course and presentation of CIDP is very variable and can be chronic progressive or relapse remitting. Electrophysiological testing reveals slowing and conduction block as indicative of an acquired demyelinating neuropathy (England & Asbury, 2004; Said, 2006). Reports of activity-dependant conduction block in CIDP could be functionally important as paresis can increase with repeated use (Pollard, 2002). Relapsing forms of CIDP tend to have a better prognosis than the more progressive types, but the degree of secondary axonal loss is most predictive of outcome.
Unlike some other polyneuropathies, CIDP is not length dependant, i.e. the longer nerves more affected. It is primarily a motor neuropathy that can affect distal and proximal muscles, though the distal ankle muscles are often the weakest (Westblad et al., 2009). Sensory impairment can also be present with some variants presenting with sensory ataxia. Symmetrical weakness and sensory changes develop in the limbs over more than 8 weeks, so differentiating from a diagnosis of GBS (Hughes, 2008). The majority of people have absent tendon reflexes and 4–15% can present with facial palsy (Said, 2006).
People with CIDP report problems with mobility and balance, but studies exploring the impact of the physical impairments are limited. In a study describing physical functioning, nearly 50% of subjects (n=21) reported that balance and walking difficulties were the worst consequences of having CIDP (Westblad et al., 2009). The same study found deficits in balance scores, timed up and go test, manual dexterity and the physical functioning domains of the SF-36 health status questionnaire.
Fatigue is reported as severe in people with CIDP and is one of the three most disabling symptoms in the majority (Merkies et al., 1999). The level of fatigue does not correlate with impairment measures of muscle strength and sensation or disease severity, so the cause of fatigue is not yet clear. The consequence of fatigue in this population, however, is the potential for a reduction in daily activity and secondary deconditioning.
Management of chronic inflammatory demyelinating polyradiculoneuropathy
Beneficial effects on the presentation of CIDP have been shown with corticosteroids, intravenous immunoglobulins and plasma exchange (Brannagan, 2009), but this is necessary over a prolonged period, which can be problematic. Long-term steroid use has difficult side-effects, IVIg is expensive and plasma exchange is inconvenient for the patient (Hughes, 2008). The response to treatment will be determined by factors such as the degree of axonal loss. Evaluating the degree of improvement can also be hampered by a poor correlation between electrophysiological data and a lack of good, objective outcome measures (Said, 2006).
Charcot-Marie-Tooth disease type 1 (CMT1)
The most common hereditary neurological condition is a polyneuropathy called Charcot-Marie-Tooth disease (CMT) causing degeneration of the peripheral nerves and affecting about 36 in a 100 000 people (Krajewski et al., 2000; Pareyson et al., 2006). The name originates from the three neurologists who described it in 1886. The first gene causing the CMT phenotype was mapped in 1989. Since then many more have been identified and the common CMT phenotype can be caused by as many as a 100 gene mutations (Nicholson, 2006). The most common types of CMT show a slow decline in distal muscle strength and sensation that predominantly affects the longer peripheral nerves. Most genes causing CMT have autosomal dominant inheritance, though there are rarer autosomal recessive types. Family history is an important step in deciding if and how a neuropathy is inherited.
Type 1 CMT (CMT1) presents with demyelination of the more thickly myelinated, fast-conducting axons, for example the alpha motor neurons and 1a afferent sensory neurons. As with other demyelinating polyneuropathies, axonal loss occurs with prolonged demyelination. In CMT 1 there is an increase in the motor unit size, that is, an increase in the number of muscle fibres supplied by one motor axon. This occurs during the chronic, slow denervation process when unaffected motor axons are able to produce collateral sprouts that re-innervate previously denervated muscle fibres (Ericson et al., 2000).
CMT 1a refers to a type of CMT1 that is caused by the most common gene mutation. It accounts for up to 80% of people with CMT and is caused by the mutation of the PMP22 gene on the short arm of chromosome 17. Duplication of this gene affects the stability of the myelin sheath causing progressive demyelination (Pareyson et al., 2006).
Presentation of Charcot-Marie-Tooth disease type 1
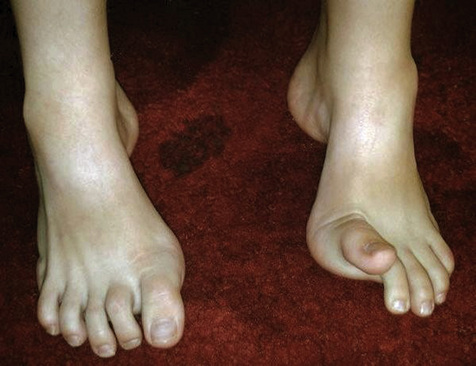
Muscle wasting is one of the key signs described for people with CMT with the classic ‘inverted wine bottle’ appearance of the distal lower limb and ‘claw hand’ of the upper limb (Figure 9.4). Magnetic resonance imaging (MRI) reveals that atrophy of the distal lower limb muscles can occur even when an individual appears unaffected on clinical examination (Gallardo et al., 2006). The distal lower and upper limb muscles tend to weaken first showing a slow decline in strength over decades. The degree and extent of weakness have been correlated with axonal loss rather than demyelination in studies of the hand (Videler et al., 2008). The proximal limb muscles are less affected but some studies have found they are still weak compared to normative data (Carter et al., 1995).
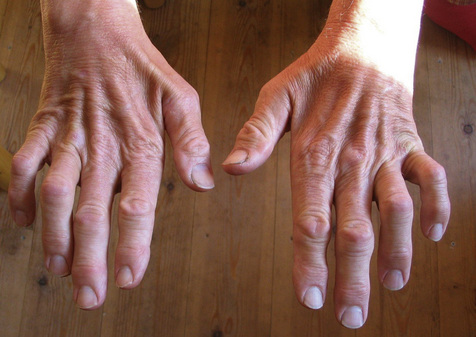
In addition to weakness and wasting, a length-dependant gradual loss of sensation occurs. People with CMT1 show a principal impairment of the thickly myelinated large diameter sensory nerves that mediate the sensations of light touch and vibration (Nardone et al., 2000). However, sensations conveyed by smaller diameter fibres, e.g. pain, temperature or pin prick, may also be reduced (Carter et al., 1995).
Little has been written about hand deformity in people with CMT, but there has been interest in the presentation and evolution of foot deformity. Pes cavus is a common foot posture observed in people with CMT1 where the condition starts early in life and is described as a foot type with an excessively high longitudinal arch and foot supination (Figure 9.5). Pes cavus is often associated with hind foot varus, toe clawing and dorsiflexion of the metatarsophalangeal joints (MTPJs). These deformities are thought to evolve through muscle imbalances over time (Guyton & Mann, 2000; Holmes & Hansen, 1993; Mann & Missirian, 1988). In support of this theory, a significant correlation was found between a pes cavus foot structure and ankle dorsiflexion range in children with CMT (Burns et al., 2009).
Exploration of upper limb function has revealed that people with CMT can have impaired manual dexterity and upper limb functional tasks (Burns et al., 2008; Videler et al., 2007; Videler et al., 2008) that are related to muscle weakness (Selles et al., 2006). In the lower limb, distal weakness has been related to foot drop and failure of the plantarflexors (Vinci & Perelli, 2002), which influences the pattern of gait as people with CMT walk. Gait analysis has revealed primary distal gait impairments with problems with foot clearance during swing, and reduced contribution of the plantarflexor muscles to progression of the trunk and swing leg (Newman et al., 2007). Further exploration revealed that people with CMT utilize additional movements of the proximal joints during walking to compensate for the primary impairments of distal weakness and sensory loss (Don et al., 2007; (Ramdharry et al., 2009).
Fatigue is well documented in people with CMT and 67% report severe fatigue (Kalkman et al., 2005). Self-reported fatigue has been shown to have an impact on specific functions, such as walking (Ramdharry et al., 2009), so there has been a lot of recent interest in the causes. Investigations of fatigue mechanisms have found that central fatigue exists, though no greater than healthy subjects, and there is a significant element of central activation failure that could be due to a long-standing reduction in sensory feedback (Kalkman et al., 2008; Schillings et al., 2007). As a group, people with CMT have been found to be less active than the general population (Kalkman et al., 2007) and are deconditioned, as measured by oxygen uptake during exercise (Carter et al., 1995). Aerobic deconditioning and disuse muscle atrophy are a likely consequence of reduced activity levels, which may also impact on fatigue and prolonged performance of daily tasks.
People with CMT complain of more pain than the general population (Padua et al., 2008a) though it is unclear whether the pain is directly due to the neuropathy or secondary musculoskeletal deformities. Burns et al. (2005a) found increased reports of pain in people with CMT who had a pes cavus foot deformity suggesting that musculoskeletal alignment may be a cause of foot pain.
Problems with balance are reported in the clinic by people with CMT. There have been limited investigations into balance impairments but reduced balance scores have been seen in children with CMT (Burns et al., 2009).
Investigations of quality of life for people with CMT have found lower scores than the general population and similar to those reported for people with stroke and other disabilities (Pfeiffer et al., 2001; Vinci et al., 2005). The reason for this is multi-factorial and measures of ambulation and axonal loss correlate with quality of life measures (Padua et al., 2008b). An interesting link has also been observed between quality of life and occupation in a study of 121 people. Using the short-form 36 measure, investigators found lower scores for physical functioning, physical role, emotional role and mental health for people who did not work (Vinci et al., 2005). It is unclear whether working improves these domains or whether the most physically, emotionally and mentally well people are able to continue to work for longer.





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


