Fig. 1
Concentration response curve of 6-OHDA on C6 glioma cell viability. The curve was generated to identify the EC50 value for the toxin on cell survival. Data are means ± SE of at least six independent experiments and are expressed as a percentage of the untreated control group
3.2 Protective Effect of Guanosine on 6-OHDA-Mediated Toxicity
C6 glioma cells were treated with 30 μM 6-OHDA for 24 h in the presence or absence of 300 μM GUO. The concentration of the guanine nucleoside was chosen on the basis of our previous results showing its neuroprotective effects in different cell types, including glial and neuroblastoma cells (Di Iorio et al. 2004; Pettifer et al. 2004). The addition of 30 μM 6-OHDA led to the expected reduction in C6 glioma cell viability (Fig. 2). GUO (300 μM) added along with the toxin (co-treatment) effectively attenuated (by about 60 %) the 6-OHDA-induced cytotoxicity as determined by MTS reduction assay (Fig. 2).
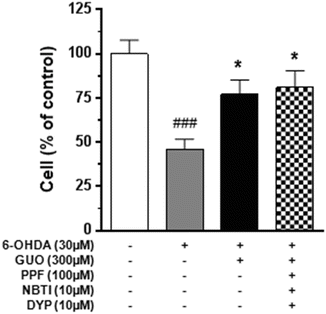
Fig. 2
Effect of guanosine (GUO) on C6 glioma cell cytotoxicity induced by 6-hydroxydopamine (6-OHDA). The effect of 300 μM guanosine on the toxicity induced by 30 μM 6-OHDA for 24 h was evaluated in the presence or absence of nucleoside transporter blockers. The blocker cocktail [10 μM 6-[(4-nitrobenzyl)thio]-9-β-D-ribofuranosylpurine (NBTI), 100 μM propentofylline (PPF), and 10 μM dipyridamole (DYP)] was added to the culture medium 1 h before 6-OHDA/GUO co-treatment. The C6 cell viability was determined by MTS assay as described in Methods. Each column represents the mean ± SE of at least six independent experiments. ###P < 0.001 vs. untreated cells (control); *P < 0.05 vs. 6-OHDA-treated cells
To evaluate whether the protective effects of GUO were mediated by intracellular mechanisms following the nucleoside uptake, the cells were pretreated for 1 h with a cocktail of known nucleoside transporter blockers (10 μM NBTI, 100 μM PPF, and 10 μM DYP) (Parkinson et al. 2006). As shown in Fig. 2, the protective effect of GUO was not affected by cell pretreatment with these drugs.
3.3 6-OHDA -Induced Apoptotic Death in C6 Glioma Cells
To determine the capability of the chosen 6-OHDA concentration to cause apoptosis, DNA-sensitive dye Hoechst 33258 staining and DNA fragmentation assay by oligonucleosomal ELISA were used. The Hoechst 33258 staining was used to assess changes in nuclear morphology following cell treatment with 30 μM 6-OHDA for 24 h in the presence or absence of 300 μM GUO. As shown in Fig. 3A, nuclei in normal C6 glioma cells exhibited diffused Hoechst 33258 staining of chromatin. In contrast, about 27 % of nuclei in cells treated with 6-OHDA showed condensed chromatin (Fig. 3A, B). No protection was reported when 300 μM GUO was added to the cell medium along with the neurotoxin.
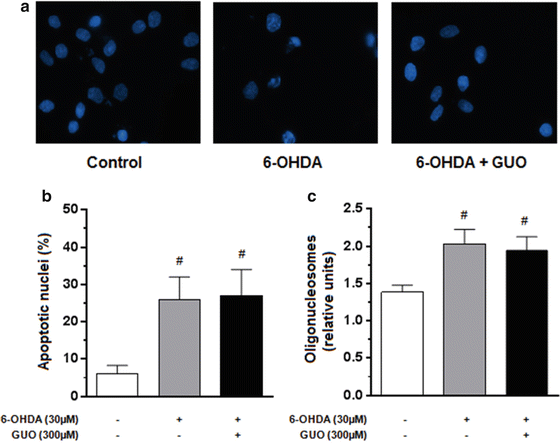
Fig. 3
Effect of 6-hydroxydopamine (6-OHDA) alone or in combination with guanosine (GUO) on C6 glioma cell apoptosis. Cultured cells were exposed to 30 μM 6-OHDA for 24 h in the presence or absence of 300 μM GUO. Untreated (control) and treated cells were stained with the fluorescent nuclear dye Hoechst 33258 (Panel A) and the statistical analysis of apoptotic cells was reported (Panel B). The apoptosis was also evaluated measuring the oligonucleosome formation by ELISA assay (Panel C). Photomicrographs are representative results taken from ten different fields from randomly selected slides. Each column value represents the mean ± SE of six independent experiments. #p < 0.05 vs. untreated cells (control)
3.4 ERK and PI3K/Akt Pathways Involvement in Neuroprotection by Guanosine Against 6-OHDA-Mediated Toxicity in C6 Glioma Cells
It has been reported that GUO induces a rapid increase in ERK1/2 and Akt phosphorylation in different cell types including PC12, microglia, and astrocytes (D’Alimonte et al. 2007; Di Iorio et al. 2004; Pettifer et al. 2004). To evaluate the involvement of the above mentioned cell survival pathways in GUO-mediated neuroprotection, we examined the effect of LY294002 (30 μM) an inhibitor of phosphoinositide-3-kinase (PI3K), an upstream of Akt, and U0126 (10 μM), an inhibitor of mitogen-activated protein kinase (MEK1/2), in glioma cell cultures co-treated with 30 μM 6-OHDA for 24 h. As shown in Fig. 4, both LY294002 and U0126 significantly reduced the effect of GUO on 6-OHDA-induced cytotoxicity as determined by MTS reduction assay. Consistent with the results obtained by MTS reduction assay, Western blotting analysis showed that 300 μM GUO was able to increase p-ERK1/2 and p-Akt – immunoreactivity in C6 glioma cells treated with the nucleoside for 15 min (Fig. 5A, C). As already reported for other cell types, including C6 glioma cells (Lee et al. 2011), also 6-OHDA increased p-ERK immunoreactivity (Fig. 5A).
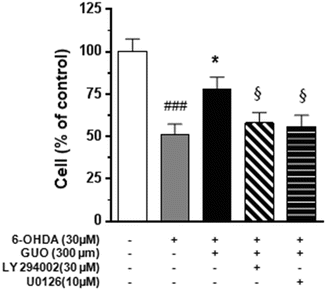
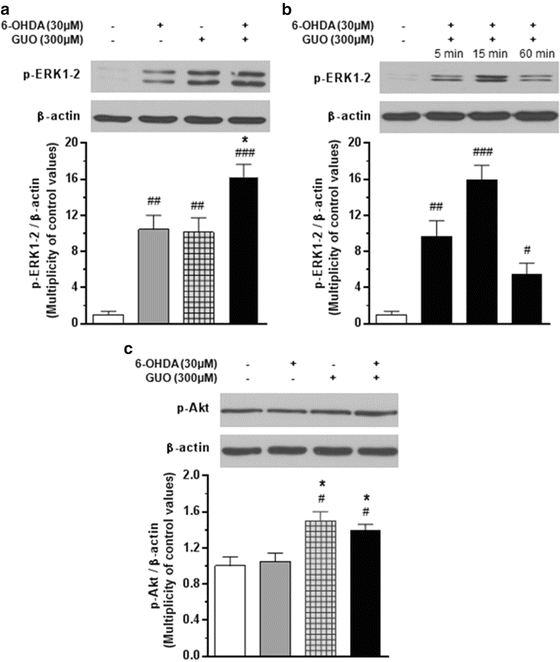
Fig. 4
Effects of PI3-kinase inhibitor (LY294002) and MEK1/2 inhibitor (U0126) on the protective effect of 300 μM guanosine (GUO) on 6-hydroxydopamine (6-OHDA)-induced toxicity. LY294002 (30 μM) or U0126 (10 μM) were added 1 h prior to the co-treatment with 300 μM GUO plus 30 μM 6-OHDA for 24 h. The C6 cell viability was determined by MTS assay as described in Methods. Each column represents the mean ± SE of at least six independent experiments. ###P < 0.001 vs. untreated cells (control); *P < 0.05 vs. 6-OHDA-treated cells; §P < 0.05 vs. the 6-OHDA/GUO co-treatment
Fig. 5
Effect of 6-hydroxydopamine (6-OHDA), guanosine (GUO), or the combined treatment on the phosphorylation of ERK1/2 (Panels A & B) and Akt (Panel C) in C6 glioma cells. Cultured cells were incubated for 15 min with 30 μM 6-OHDA, 300 μM (GUO) or 6-OHDA/GUO co-treatment and harvested for western blot analysis. The protein expression of phosphorylated ERK1/2 (p-ERK1/2) or Akt (p-Akt), and β-actin were determined by using specific antibodies as described in Methods. To evaluate the time course of ERK1/2 activation induced by the co-treatment, cells were incubated for 5, 15, and 60 min with 30 μM 6-OHDA plus 300 μM GUO (Panel B). For each signaling pathway, a representative immunoblot is shown. Each column represents the mean ± SE of three independent experiments. #P < 0.05, ##P < 0.01, ###P < 0.001 vs. untreated cells (control); *P < 0.05 vs. 6-OHDA treated cells
It has been reported that sustained activation of ERK1/2 does not protect against either L-DOPA- or 6-OHDA-induced cytotoxicity (Jin et al. 2010). Thus, in an attempt to better define the characteristics of ERK activation mediated by co-treatment of C6 glioma cells with 30 μM 6-OHDA plus 300 μM GUO, analysis of the kinetics of ERK phosphorylation was performed. As shown in Fig. 5B, co-treatment significantly increased p-ERK1/2 level within 5 min, peaking at 15 min after administration, and strongly decreased it by 60 min.
Contrary to what was found for ERK1/2 phosphorylation, the neurotoxin was unable to enhance the levels of p-Akt in the same cells (Fig. 5C), whereas following the co-treatment with GUO and 6-OHDA, p-Akt levels increased by 39 % compared with the control values. The effect of cell co-treatment with 300 μM GUO and 30 μM 6-OHDA on p-ERK1/2 and p-Akt immunoreactivity was not affected by C6 glioma cell pretreatment with the cocktail of the nucleoside transporter blockers reported above (10 μM NBTI, 100 μM PPF, and 10 μM DYP) (Fig. 6A, B).
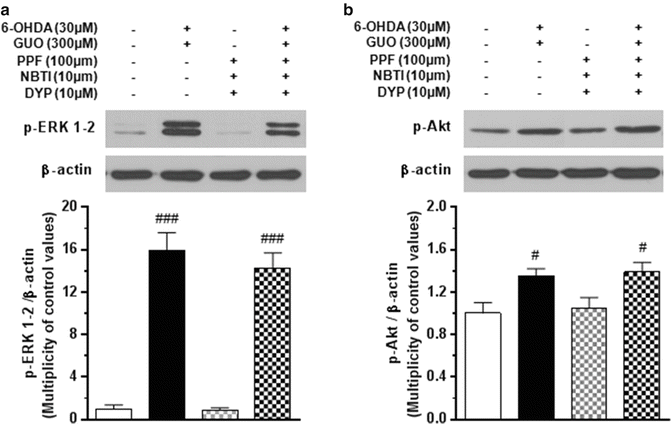
Fig. 6
Effect of the nucleoside transporter blockers on the phosphorylation of ERK1/2 (Panel A) and Akt (Panel B) induced by the C6 cell co-treatment with 6-hydroxydopamine (6-OHDA)/guanosine (GUO). Cultured cells were incubated for 15 min with 30 μM 6-OHDA plus 300 μM GUO. When used, the blocker cocktail [10 μM 6-[(4-nitrobenzyl)thio]-9-β-D-ribofuranosylpurine (NBTI), 100 μM propentofylline (PPF), and 10 μM dypiridamole (DYP)] was added to the culture medium 1 h before 6-OHDA/GUO cotreatment. The protein expression of phosphorylated ERK1/2 (p-ERK1/2) or Akt (p-Akt) and β-actin were determined by using specific antibodies as described in Methods. For each signaling pathway, a representative immunoblot is shown. Each column represents the mean ± SE of three independent experiments. #P < 0.05, ###P < 0.001 vs. untreated cells (control)
4 Discussion
In the present study we demonstrate that 6-OHDA, a neurotoxin used to induce experimental models of PD, caused cytotoxicity in a concentration-dependent manner in C6 glioma cells, taken as a model system for astrocytes. We also report that 300 μM GUO concomitantly administered with 6-OHDA counteracted the toxin-induced loss of viability in this in vitro model of PD. It has been shown that GUO and its metabolic product guanine are taken up into both neurons and astrocytes mainly via the equilibrative nucleoside transporter (Parkinson et al. 2006). Thus, to determine whether GUO-mediated cytoprotection was due to intracellular effects following its uptake by the cells, a cocktail of uptake inhibitors was used. In agreement with previous findings observed in primary cultures of astrocytes (Di Iorio et al. 2004), the results show that the transport of GUO and its consequent intracellular accumulation was not required for protecting the cells in our in vitro model of PD. This fits well with our previous data which pointed to the presence of specific binding sites for GUO on rat brain membranes (Traversa et al. 2002, 2003).
The mechanisms underlying neurodegenerative processes in PD are still unclear. However, increasing evidence indicates that apoptosis is involved in the loss of dopaminergic neurons. We recently reported that GUO (300 μM) protected SH-SY5Y neuroblastoma cells when they were exposed to 6-OHDA, promoting their survival by (i) reducing the neurotoxin-mediated activation of p-38 and JNK; (ii) causing an early increase in phosphorylation of the anti-apoptotic kinase Akt; (iii) inactivating the pro-apoptotic factor GSK3β; and (iv) increasing the expression of the anti-apoptotic Bcl-2 protein (Giuliani et al. 2012b).
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


