Psychiatric and Cognitive Phenomena as an Expression of Epileptic Encephalopathies
Andres M. Kanner
Ruben Kuzniecky
The classification of psychiatric clinical phenomena in patients with epilepsy is frequently based on their temporal relation with seizure occurrence. Hence, we refer to ictal psychiatric symptoms when symptoms are the clinical expression of the actual seizure; peri-ictal psychiatric symptoms refer to symptomatology preceding and/or following the ictus (i.e., postictal); and interictal symptoms are those that present independently of the seizure occurrence. A pathogenic role of the epileptic process on psychiatric phenomena is obvious in peri-ictal disorders, but to a significantly lesser degree in interictal disorders. Yet, at some point in their practice, all epileptologists have encountered cases of patients with florid “interictal” psychiatric and cognitive disturbances that become apparent around the beginning of a seizure disorder and remit and/or significantly improve upon its elimination. We use the term epileptic encephalopathies to refer to this type of psychiatric and cognitive disturbances. The purpose of this chapter is to review the available data on two epileptic encephalopathies: (i) the acquired epileptic aphasia of childhood (also known as Landau-Kleffner Syndrome [LKS]), in which the “nonepileptic” clinical disturbances are the major expression of the epileptic disorder, and cure of the seizure disorder is often followed by partial to full recovery of language function (1) and (ii) the epileptic encephalopathy associated with hypothalamic hamartomas with gelastic epilepsy (HHGE) presenting characteristically with severe psychiatric and cognitive disturbances associated with the onset of a variety of epileptic seizures, particularly gelastic seizures, and also including complex partial, secondary generalized tonic-clonic (GTC) seizures, and at times atonic or tonic seizures (2). These severe psychiatric and cognitive disturbances remit or improve significantly with the cessation of the seizures following a successful surgical treatment.
As shown subsequently, the severity of the actual clinical seizures may range from very rare or nonexistent such as in LKS but with electroencephalographic (EEG) recordings showing abundant epileptiform activity in the form of a continuous spike and wave during slow wave sleep (CSWS) pattern. Gelastic seizures associated with hypothalamic hamartomas are more likely to be refractory to antiepileptic drugs (AEDs) and often of several types.
The underlying pathogenic mechanisms of the psychiatric and cognitive disturbances of these two epileptic encephalopathies are suspected to be closely tied to the epileptic activity and its impact on brain structures that mediate specific cognitive, affective, and behavioral functions. To date, however, the mechanisms responsible have yet to be established. In the case of LKS, for example, some of the possible culprits include (i) a direct impact of interictal spikes on cognitive functions and (ii) neuronal (and specifically synaptic) changes triggered by continuous interictal bombardment, among others. In HHGE, thalamic and subthalamic electrographic involvement by frequent ictal and interictal activity is possible. Alternatively, hypothalamic endorphin dysfunction has been implicated but never proven.
The impact of interictal epileptiform activity on cognitive functions was studied extensively by Binnie et al. (3,4). Although it is still the source of much debate, there appears to be agreement that generalized and focal discharges can often be associated with disturbed cognition. Schwab (5) in 1939 was the first to demonstrate an absent or delayed response during a generalized epileptiform discharge using a simple reaction task test. Aarts et al. (6) coined the term transient cognitive impairment to reflect this phenomenon. Binnie et al. (4) described transient cognitive impairment in 50% of 91 patients during generalized or focal discharges. Right-sided discharges were associated with impairment in visual spatial tasks, whereas errors in verbal tasks were demonstrated during left-sided discharges. The authors observed that the error rates increased if the stimulus was presented in the midst of the discharge. If the epileptiform discharge occurred within 2 seconds before the stimulus, the degree of disruption was greater. Kasteleijn-Nolst Trenite et al. (7) reported similar findings with respect to the type of deficit in relation to the side of discharge in 36% of a group of 69 children. The relationship between the occurrence of epileptiform discharge and cognitive functions is complex, as increased concentration on a task can result in a decrease in the rate of epileptiform discharges (8).
With respect to the second potential mechanism, Grigonis and Murphy (9) demonstrated disruption in the formation of normal synaptic connections by focal epileptic discharges in the brain of young animals. Along the same lines, Morrell attributed the pathogenic mechanisms mediating language disturbances in LKS to the interference of the normal process of synaptic organization in language cortex by the constant “bombardment” of interictal epileptic activity (10,11): “It is well known that different neural systems have critical periods in development during which the adult patterns of synaptic engagement are gradually established. At such times, there is first a superabundant outgrowth of axon terminals resulting in a temporary hyper-innervation of the synaptic target; this extravagant outgrowth appears to be entirely under genetic control. Over the remainder of the critical period, the synaptic contacts are extensively pruned. The pruning is selective; it is dependent on and controlled by synaptic use, i.e., environmental input. Synaptic contacts activated by use become cemented, so to speak, while those not environmentally engaged wither away (12,13,14,15,16,17,18). Neural networks for linguistic function develop late and their circuitry remains malleable during the first 8 to 10 years of life (19,20,21,22). We hypothesize, then, that the impact of epileptic activity originating from this anlage of speech cortex may result in chaotic and behaviorally meaningless activation of synaptic contacts that would have been ‘pruned’ under normal circumstances. Such epileptic activity may set up permanent, inappropriate and nonfunctional linkages.”
Acquired Epileptic Aphasia of Childhood
Acquired epileptic aphasia of childhood (or LKS) is an acquired epileptic aphasia or verbal auditory agnosia occurring in children who have already developed age-appropriate language function. It is thought
to result from an epileptogenic lesion arising in the speech cortex during a critical period of development. It was first described in 1957 by Landau and Kleffner in a report on six patients (1). In 1985, LKS was included in the International Classification of Epileptic Syndromes, giving it legitimacy as a recognized clinical entity.
to result from an epileptogenic lesion arising in the speech cortex during a critical period of development. It was first described in 1957 by Landau and Kleffner in a report on six patients (1). In 1985, LKS was included in the International Classification of Epileptic Syndromes, giving it legitimacy as a recognized clinical entity.
LKS has a clearly defined set of clinical and electrographic characteristics. These include a receptive speech disturbance or verbal auditory agnosia (23), followed soon after by disturbances of expressive speech, which can evolve to a state of complete mutism. The onset of speech disturbance coincides with that of seizure activity (see following text).
The symptoms of LKS appear between 2 and 8 years of age, characteristically with an acute or subacute presentation, although in some children the onset can have a stuttering course (24,25). The initial linguistic disturbances include problems with verbal comprehension (verbal auditory agnosia) that can often be mistaken for acquired deafness (26). Soon after, speech output is affected and paraphasias and phonological errors appear. Symptoms may progress in a steady or fluctuating manner to a state of complete mutism, during which the child will fail to respond even to familiar nonverbal sounds, such as the ring of the doorbell or the telephone, or the dog barking. The speech disturbances are associated with behavioral changes, which may include motor hyperactivity in up to 50% of children (24,25) and sleep disturbances. At the height of the auditory agnosia, some autistic-like features, such as self-stimulatory behavior, may be identified. However, the child with LKS never loses the ability to relate with family members and understand social cues. The onset of the language disorder is concurrent with that of seizures and/or electrographic evidence of epileptiform activity presenting characteristically, but not exclusively, as spike-wave discharges with a bilateral distribution, maximal in the posterior temporal regions of each hemisphere. This EEG pattern may occupy more than 80% of slow wave sleep, and is known as CSWS (Fig. 21.1) (24).
Twenty percent to 30% of children will never exhibit any evidence of clinical seizures (24). Seizures are often nocturnal; their clinical phenomena vary widely and are often very subtle. In our experience, seizures are identified for the first time in the course of prolonged video electroencephalographic (VEEG) monitoring study in some patients. The clinical phenomena include brief clonic deviation of the eyes, at times associated with eye blinking, head dropping, and minor automatisms with occasional secondary generalization (27). Seizures, unlike the language disorder, respond readily to AED therapy (24,25) and generally subside by the age of 15 (28). Another important diagnostic criterion of LKS is the relative isolation of the behavioral deficit to the linguistic and auditory perceptual spheres, that is, children show relatively normal performance on nonverbal cognitive tasks (27).
In some cases, spontaneous remission may occur weeks or months after onset (26). Mantovani and Landau (29) reported a considerable degree of recovery in four of nine patients reevaluated 10 to 28 years after the onset of their symptoms. Deonna et al. (30) followed seven patients diagnosed with LKS into their young adult years. Only one patient recovered completely; a second patient had normal language but was severely dyslexic; a third patient had only recovered comprehension; and the other four patients showed lack of comprehension and continued displaying severe expressive language problems. Review of the available data suggests that when symptoms persist unchanged for more than 1 year, spontaneous recovery is rare, and a severe lifelong linguistic handicap is the common result (25,26,31,32,33,34,35).
Figure 21.1 illustrates the classic EEG tracing of CSWS recorded from a child with LKS. Despite the bilateral distribution of this epileptiform activity, a unilateral source can be identified in many of these children by using the methohexital suppression test (27) or with magnetoencephalography (MEG) (36). A rather consistent distribution of spikes can be identified with referential montages
and field potential mapping, consisting of a frontal negativity and a temporal positivity, (37) and reflects a generator source represented by a tangential dipole in the inferior bank of the sylvian fissure (on the superior surface of the temporal lobe). We refer to this pattern as the sylvian dipole. The presence of this sylvian dipole has also been demonstrated with MEG (36) (Fig. 21.2).
and field potential mapping, consisting of a frontal negativity and a temporal positivity, (37) and reflects a generator source represented by a tangential dipole in the inferior bank of the sylvian fissure (on the superior surface of the temporal lobe). We refer to this pattern as the sylvian dipole. The presence of this sylvian dipole has also been demonstrated with MEG (36) (Fig. 21.2).
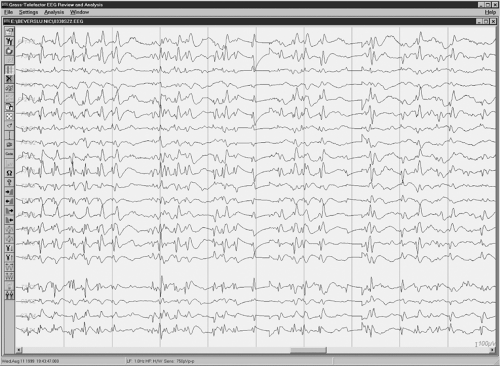 Figure 21.1 Electrographic pattern of continuous spike and wave during slow wave sleep in a child with Landau-Kleffner syndrome before surgical treatment with multiple subpial transection. |
Several authors have suggested a possible relationship between the severity of linguistic deficits, behavioral abnormalities, and EEG abnormalities. This relationship is supported by the significant improvement in both domains with the normalization of EEG recordings during drug trials with steroids and by the recurrence of symptoms and epileptiform discharges on electroencephalogram following their discontinuation (38,39,40,41,42).
As stated earlier, LKS can remit spontaneously in a number of children. In those with persistent symptoms, pharmacological treatment with AED has failed to yield encouraging results. Certain AEDs, such as carbamazepine, can worsen the severity of language dysfunction and behavior. Valproic acid and clonazepam are the two AEDs most widely used in LKS, but their efficacy is limited at best when symptoms have taken a chronic course. Steroids can yield a significant improvement of language deficits, together with eradication of the epileptic pattern of CSWS in approximately 50% of children. It should be noted, however, that in some of these children, such eradication of epileptic activity fails to exert any impact on language deficits or behavioral problems. The lack of a consistent relationship between eradication of spikes and symptom remission in some of these children is puzzling and is yet to be understood. The acute and long-term side effects of steroids limit the duration of therapy.
Morrell et al. (27) suggested a surgical treatment for LKS with the use of
multiple subpial transection (MST) and, in 1995, reported their results of the first 14 children treated surgically with this technique. This surgical technique was developed with the aim of eliminating epileptic activity from eloquent cortex without interfering with function (43). The technique interrupts intracortical horizontal fibers that are necessary for the generation of epileptic discharges (44,45,46,47), while preserving the vertical columnar organization of the cortex, the basic functional unit of cortical physiology (48,49,50).
multiple subpial transection (MST) and, in 1995, reported their results of the first 14 children treated surgically with this technique. This surgical technique was developed with the aim of eliminating epileptic activity from eloquent cortex without interfering with function (43). The technique interrupts intracortical horizontal fibers that are necessary for the generation of epileptic discharges (44,45,46,47), while preserving the vertical columnar organization of the cortex, the basic functional unit of cortical physiology (48,49,50).
Related posts:
 Animal Models of Depression and Epilepsy: The Genetically Epilepsy-Prone Rat
Psychiatric Aspects of Pediatric Epilepsy
The Psychoses of Epilepsy
Attention-Deficit Hyperactivity Disorder, Attention Problems, and Epilepsy
Psychiatric Issues in Patients with Epilepsy and Mental Retardation
Animal Models of Depression and Epilepsy: The Genetically Epilepsy-Prone Rat
Psychiatric Aspects of Pediatric Epilepsy
The Psychoses of Epilepsy
Attention-Deficit Hyperactivity Disorder, Attention Problems, and Epilepsy
Psychiatric Issues in Patients with Epilepsy and Mental Retardation
 Health Outcomes in Epilepsy: The Influence of Psychiatric Comorbidity and Other Factors
Health Outcomes in Epilepsy: The Influence of Psychiatric Comorbidity and Other Factors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


