A. Development of multiple cognitive deficits
A1. Memory problems (e.g., amnesia)
A2. At least one or more of the following:
Aphasia (language impairment)
Apraxia (loss of the ability to execute learned purposeful movements despite intact physical abilities)
Agnosia (loss of the ability to recognize objects, persons, sounds, shapes, or smells despite an intact sensory system)
Impairment in executive functioning (e.g., organizing, abstract reasoning)
B. Items described in A1. and A2. cause a significant decline in social and/or occupational functioning compared to a previously higher level of functioning
C. The cognitive deficits in A1. and A2. are not due to:
C1. Other diseases of the central nervous system (e.g., brain tumors, cerebrovascular accidents)
C2. Systemic disorders (e.g., vitamin B12 deficiency, hypothyroidism)
C3. Substance-related diseases
D. Diagnosis of dementia is not applicable if cognitive dysfunctioning occurs exclusively during the course of a delirium
E. Cognitive deficits are not attributed to another Axis I disorder such as depression or schizophrenia
Besides the cognitive aspects, dementia is also characterized by numerous behavioral symptoms entitled Behavioral and Psychological Signs and Symptoms of Dementia (BPSD) (Reisberg et al. 1987). BPSD consist of delusional ideation, hallucinations, activity disturbances, agitation/aggression, circadian rhythm disturbances, affective disturbances, and anxiety disorders and are considered a major component of the dementia syndrome. Lastly, basic (BADL) and instrumental activities of daily living (IADL) complete the definition of dementia. BADL refer to daily self-care activities such as personal hygiene, getting dressed, eating, and general mobility, whereas IADL require more complex abilities such as driving a car, utilizing a phone, taking medication, doing groceries, and managing finances (Lawton and Brody 1969). During the course of dementia, IADL are firstly affected and are later followed by BADL (Gauthier et al. 1997). Several studies also showed a direct association between cognitive decline and worsening of BADL and IADL in dementia patients and non-demented elderly (Mitnitski et al. 1999).
The definition above emphasizes that the term “dementia” is a syndrome (i.e., association of several clinically recognizable features, signs, and symptoms) rather than only a cognitive disorder and is completed by important behavioral and functional shortcomings as well (Fig. 11.1).
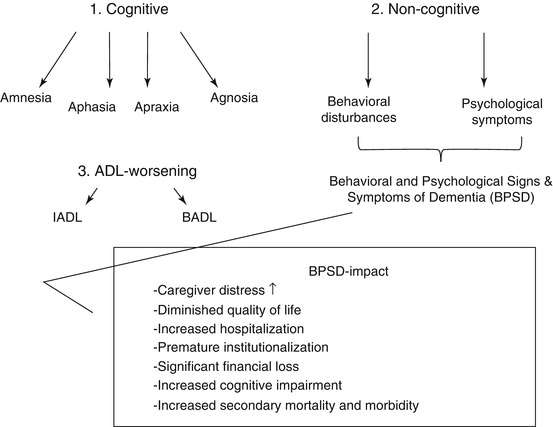
Fig. 11.1
The dementia syndrome consists of cognitive and noncognitive symptomatology. Worsening of BADL and IADL completes the definition. BPSD examples are delusional ideation and hallucinations, activity disturbances, aggression and agitation, sleep disturbances, mood disorders, and anxiety. Abbreviations: BADL basic activities of daily living, BPSD Behavioral and Psychological Signs and Symptoms of Dementia, IADL instrumental activities of daily living
11.1.2 Prevalence and Incidence
Although dementia strikes irrespective of age, the prevalence of dementia generally rises with it. Women seem to be more frequently affected by dementia than men (Breteler et al. 1992) although this observation might be attributed to a slower progression rate of the disease in women combined with a proportionally longer life expectancy (Bachman et al. 1993). Prevalence estimates of dementia in the aged population show distinct variation due to differences in population selection, case ascertainment procedures, and diagnostic criteria, which often results in over- or underestimation of dementia occurrence (De Deyn et al. 2011). In general, however, the prevalence of moderate to severe dementia approximately doubles every 5 years starting at a rate of 2 % between the age of 65 and 69, augmenting to 4 % in people aged between 70 and 74 up to 16 % in octogenarians (Henderson 1990; Morris 1994). These numbers correspond to a prevalence of 5 up to 10 % in the elderly aged 65 and older. In Europe, the prevalence of dementia varies between 1 % at the age of 60–64 rising up to 34.7 % in elderly aged 95–99 (Hofman et al. 1991). In the Netherlands, prevalence of dementia in people aged 75–79 was estimated to be 5.2 % in 1992 (in a rural area near Zwolle) (Boersma et al. 1998) and 6.1 % in 1993 (in the Rotterdam suburb of Ommoord) (Ott et al. 1995; Breteler et al. 1998), while in Belgium, it was estimated to be 7.6 % in 1993 (in the semirural area of Heist-op-den-Berg) (Roelands et al. 1994). More recent figures of Belgian dementia prevalence estimates came from the Antwerp Cognition (ANCOG) study. This longitudinal cohort study of 825 community-dwelling elderly aged between 75 and 80, living in 6 different districts of Antwerp, with a 3-year follow-up period (n = 363) resulted in an overall prevalence rate of 8.7 % (De Deyn et al. 2011).
To give exact numbers, Wimo et al. (2003) assessed the worldwide occurrence of dementia from 1950 until 2000 and also estimated its progression until 2050. The worldwide number of persons with dementia in 2000 was estimated at about 25 million persons. Almost half of the demented individuals lived in Asia (46 %), 30 % in Europe, and 12 % in North America. Fifty-two percent lived in developing regions. About 6.1 % of the population aged 65 years and older suffered from dementia (about 0.5 % of the worldwide population) and 59 % were female. The number of new cases of dementia in 2000 was calculated to be approximately 4.6 million. The forecast indicated a considerable increase in the number of demented elderly from 25 million in the year 2000 to 63 million in 2030 (41 million in less developed regions) and to 114 million in 2050 (84 million in developing regions).
It thus becomes clear that due to progressive aging of the general population, a further increase of dementia prevalence during the next decades is expected. Moreover, the majority of demented elders live in less developed countries and this proportion will increase considerably in the future.
Less data is available regarding dementia incidence estimates (i.e., a measure of the risk to develop dementia within a specific period of time). Versporten et al. (2005) reported an overall incidence rate of dementia of 41 per 1,000 person years (Py) for men and 33 per 1,000 Py for women (i.e., 41 or 33 persons out of 1,000 that were observed for 1 year). This Epidemiology Research on Dementia in Antwerp (ERDA) study started in 1990 and consisted of 937 non-demented elderly aged 65 and older. Moreover, individuals with less than 7 years of education in this study population were – independently of gender – at higher risk of developing dementia compared with those receiving higher education (Versporten et al. 2005). Accordingly with the ERDA study, the ANCOG study resulted in a cumulative incidence rate of 36.60 per 1,000 Py with annual incidence rates ranging from 34.39 over 35.16 to 49.09 per 1,000 Py. In America, the average incidence rate varies between 3 per 1,000 Py in people aged 65 up to 69 years old and a maximum of 56 per 1,000 Py in 90-year-olds (Kukull et al. 2002). These figures are consistent with a previously executed large-scale European study (Launer et al. 1999).
11.1.3 Alzheimer’s Disease (AD) and Specific Dementia Syndromes
Dementia syndromes are commonly subdivided according to their reversible or irreversible characteristics (Katzman et al. 1988).
Primary dementia syndromes are irreversible neurodegenerative disorders such as Alzheimer’s disease (AD), frontotemporal dementia (FTD), dementia with Lewy bodies (DLB), Parkinson’s disease dementia (PDD), Huntington’s disease, and Creutzfeldt-Jakob disease.
On the contrary, secondary dementia syndromes are “potentially” reversible and originate from a specific acquired central nervous system disorder which led to “dementia-like deficits” (i.e., cognitive dysfunction, behavioral phenomenology). Some examples are brain tumors, cerebrovascular accidents (vascular dementia (VAD)), infections (meningitis, AIDS dementia complex), head traumas (subdural hematoma), alcohol abuse (Korsakoff syndrome), or normal pressure hydrocephalus.
Lastly, pseudodementias are “completely” reversible dementia subtypes that very much resemble primary dementia syndromes although the aspect of abundant neurodegeneration itself is absent. Examples are psychiatric disturbances (depression, schizophrenia), endocrine/metabolic disorders (hypothyroidism), malnutrition/vitamin deficiency (vitamin B12 or folic acid deficiency), or toxicological-/pharmacological-/substance-related conditions (certain sleep medication, anxiolytic, or sedatives) (Katzman et al. 1988).
For this chapter, we will be exclusively focusing on primary dementias such as AD, FTD, and DLB. Secondary dementia syndromes and pseudodementias will not be considered in the further discussion of this chapter.
11.1.3.1 Alzheimer’s Disease (AD)
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder and is named after Dr. Alois Alzheimer, who first described this syndrome in 1907 in a 51-year-old patient who suffered from a progressive cognitive impairment associated with behavioral changes and brain atrophy. AD (code 294.1x) applies with the DSM-IV-TR criteria for the dementia syndrome described above (Table 11.1) (American Psychiatric Association 2000) and is manifested by multiple cognitive deficits such as memory impairment but also aphasia, apraxia, agnosia, and/or executive dysfunctioning. Additionally, AD is encoded based on the presence (294.11) or absence (294.10) of an associated clinically significant behavioral disturbance.
Diagnosis
The National Institute on Aging and the Alzheimer’s Association workgroups (McKhann et al. 2011) recently updated the National Institute of Neurological and Communicative Disorders and Stroke (NINCDS) and the Alzheimer’s Disease and Related Disorders (ADRDA) criteria of 1984 (McKhann et al. 1984) which subdivided AD into probable, possible, and definite AD. Probable AD is characterized by cognitive deficits in at least 2 cognitive domains with an insidious onset and a progressive worsening over time, a clear-cut history of cognitive worsening by report or observation and the most prominent cognitive deficits are evident on history or clinical examination in an amnestic (e.g., impairment in learning recall and at least 1 other cognitive domain) or nonamnestic (aphasia/apraxia/agnosia/executive dysfunctioning) manner (core criteria) (McKhann et al. 2011). Supportive criteria are among others: a family history of AD, associated BPSD, disturbed ADL, and a CT scan not displaying central nervous system pathology which may underlie the dementia syndrome (McKhann et al. 1984). A new subcategory of probable AD, compared to the 1984 criteria, is the probable AD with evidence of the AD pathophysiological process category. In this new diagnostic entity, biomarker evidence of cerebrospinal fluid (CSF) amyloid beta (Aβ), total and phosphorylated tau levels, positive PET amyloid imaging, or a decreased 18F-fluorodeoxyglucose (FDG) uptake on PET in the temporoparietal cortex may increase the certainty of an active AD pathophysiological process in persons who meet the core clinical criteria for probable AD (McKhann et al. 2011). Patients who met the 1984 NINCDS-ADRDA criteria for probable AD would also correspond with the more recent 2011 criteria described above.
Possible AD differs from probable AD as it is manifested by a somewhat atypical course and heterogeneity of symptoms with an either sudden onset of cognitive impairment or an etiologically mixed presentation, such as concomitant cerebrovascular disease. The core criteria of AD, however, remain present (McKhann et al. 2011).
Pathophysiological Mechanisms
AD and other dementia subtypes are all proteinopathies. The histopathological hallmarks of the AD brain are extracellular deposits of Aβ plaques and intracellular neurofibrillary tangles (NFT) which lead to a widespread synaptic loss and neurodegeneration with a consequent neurotransmission failure, especially of the cholinergic neurotransmitter system (Van Dam and De Deyn 2006). Familial AD is an autosomal dominant disorder with an onset before the age of 65 (Blennow et al. 2006). A mutation in the amyloid precursor protein (APP) gene on chromosome 21 or in the presenilin 1 (PSEN1) or presenilin 2 (PSEN2) genes accounts for most of the familial cases. However, the familial form is rare with a prevalence of approximately 1 % (Harvey et al. 2003). In most sporadic AD cases (>95 %) with an age of onset above 65, the etiology is not entirely known. So far, only risk genes have been identified such as the apolipoprotein E (APOE) ε4 allele which increases the risk of the disease by three times in heterozygotes and 15 times in homozygotes (Farrer et al. 1997).
The amyloid cascade hypothesis is the most dominant etiological AD hypothesis and states that Aβ accumulation results from an imbalance between Aβ production and clearance (Blennow et al. 2006). Physiologically, APP is a cell membrane expressed protein not only in neurons but also in many other tissues and is likely to be involved in maintenance and modulation of neuronal networks (Loo et al. 1993). Posttranslational cleavage of APP by consecutive α- and ɣ-secretases releases a p3 fragment (non-amyloidogenic pathway), whereas the combined effect of β- and ɣ-secretases releases non-soluble Aβ peptides of various lengths, i.e., Aβ1-40 or Aβ1-42 (amyloidogenic pathway). In normal situations, the non-amyloidogenic pathway is mostly active. In familial AD, however, a mutation in PSEN1/PSEN2 (which form the catalytic subunits of the secretases) or around the cleavage site of APP causes an overproduction of the hydrophobic Aβ1-42 and consequently leads to a shifted Aβ1-40/Aβ1-42 balance. As a result, enormous amounts of Aβ1-42 fragments aggregate and form extracellular “senile plaques” (Hardy and Selkoe 2002) (Fig. 11.2). Whereas in familial AD, there is an overproduction of Aβ1-42 due to certain mutations, sporadic AD cases seem to fail sufficient Aβ clearance which leads to gradually increasing and accumulating Aβ levels in the brain. As mentioned above, genetic risk factors such as APOE ε4 but also aging and certain environmental risk factors were proven to be strongly associated with sporadic AD (Blennow et al. 2006).
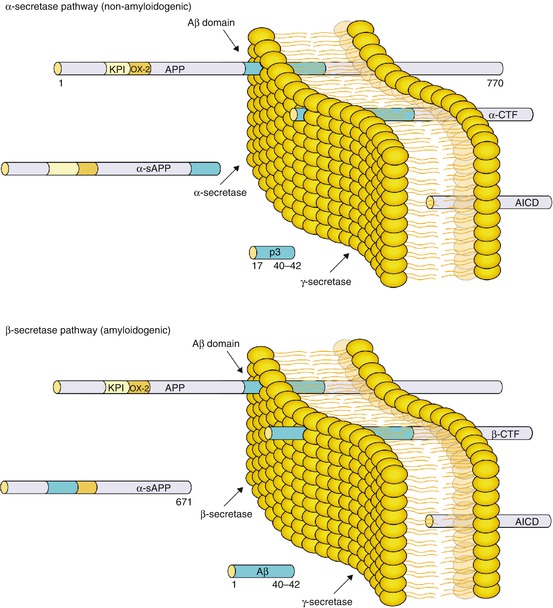
Fig. 11.2
The amyloid cascade hypothesis in AD. Amyloid precursor protein (APP) is a large transmembrane protein which is consecutively cleaved by α- and ɣ-secretases (non-amyloidogenic pathway) so that soluble p3 fragments are formed. In familial Alzheimer’s disease (AD), however, dysfunctional cleavage of APP by β- and ɣ-secretases (amyloidogenic pathway) releases larger amyloid beta (Aβ) 1–40 and 1–42 fragments which leads to an overproduction of specifically Aβ1-42. Aβ1-42 are hydrophobic, insoluble filaments that will aggregate and form amyloid “senile” plaques, the hallmark of AD pathology. In sporadic AD, Aβ aggregates are formed as well but this time due to a failed Aβ clearance. It has been suggested that an imbalance between Aβ production and clearance lies at the basis of AD pathogenesis as such. AICD APP intracellular domain, CTF C-terminal fragment, KPI Kunitz-type protease inhibitor, sAPP soluble APP (Reprinted from Blennow et al. (2006), with permission. Copyright@2006 Elsevier)
The second hallmark of AD pathology is the presence of intracellular NFT, which results from the hyperphosphorylation and aggregation of the axonal tau proteins, a group of microtubule-associated proteins that contribute to the assembly and stabilization of microtubules in neurons among others (Grundke-Iqbal et al. 1986). Tau phosphorylation is regulated by the balance between multiple kinases (e.g., GSK-3β and CDK5) and phosphatases (e.g., PP-1 and PP-2A) (Iqbal et al. 2005). An imbalance between the protein kinases and phosphatases causes tau to be hyperphosphorylated into insoluble fibrils, also called “paired helical filaments.” Tau hyperphosphorylation starts intracellularly and leads to sequestration of normal tau and other microtubule-associated proteins, which causes disassembly of microtubules and thus impaired axonal transport, compromising neuronal and synaptic function (Iqbal et al. 2005). Tau pathology starts early in the disease process in neurons of the transentorhinal region, from where it further spreads to the hippocampus and amygdala and finally to other cortical and neocortical association areas (Braak et al. 1999; Smith 2002) (Fig. 11.3).
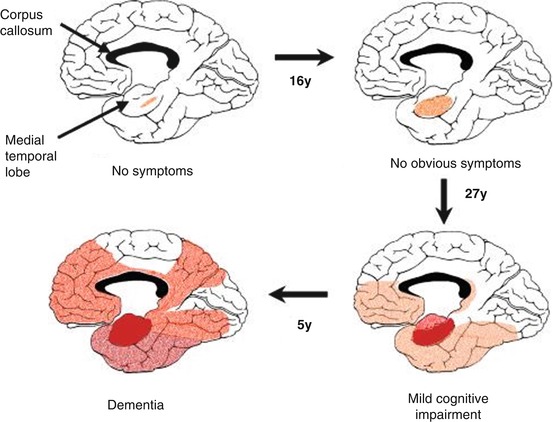
Fig. 11.3
Progressive expansion of neurofibrillary tangles (NFT) in an AD brain, showing the medial aspect of the cerebral cortex. The depth of the red color is in proportion to the density of tangles (Reprinted from Smith (2002), with permission. Copyright@2002 National Academy of Sciences, USA)
Besides Aβ deposits and NFT, oxidative stress and inflammation are two key factors in the etiological hypotheses of AD as well.
Oxidative damage to different classes of biological molecules such as sugars, lipids, proteins, and DNA is a common aspect of both normal aging and most neurodegenerative disorders (Moreira et al. 2005). In early AD, oxidative stress might have an important pathogenic role as neurons themselves use different antioxidant defense systems in case of increased oxidative stress. Evidence demonstrates that Aβ depositions and hyperphosphorylation of tau form two primary defense lines against oxidative stress. With disease progression, both Aβ and tau transform into prooxidants due to a profound redox imbalance (Smith et al. 2002).
With regard to inflammation, it has been proven that many neuroinflammatory mediators are upregulated in affected areas of the AD brain, including prostaglandins, complement components, anaphylatoxins, cytokines, chemokines, proteases, protease inhibitors, adhesion molecules, and free radicals (Akiyama et al. 2000). Côté et al. (2012) recently established a direct association between the prolonged use of nonsteroidal anti-inflammatory drugs (NSAIDs), which target cyclooxygenase (COX), and a decreased risk of subsequently developing AD even though several other clinical studies using NSAIDs in AD patients yielded a negative outcome (ADAPT Research Group et al. 2008; Breitner et al. 2011). Initially, the effect of NSAIDs in AD was thought to be attributed to a reduction of inflammation (Van Dam and De Deyn 2006). In 2001, however, it was reported that a subset of NSAIDs reduced Aβ1-42 production in cultured cells and mouse brain through a mode of action different from COX inhibition (Weggen 2001). On the other hand, the initial assumption of possible underlying anti-inflammatory mechanisms of NSAIDs in AD should not be completely abandoned (Van Dam and De Deyn 2006).
Interestingly, the induced neuroinflammation in AD might also lie at the basis of some BPSD, such as depression. For example, the enzyme indoleamine 2,3-dioxygenase (IDO) metabolizes tryptophan, the precursor of serotonin (5HT), into kynurenine. Due to neuroinflammation, the IDO activity becomes upregulated and eventually the kynurenine catabolization further leads to an overproduction of quinolinic acid, the neurotoxic end product of the tryptophan pathway which also contributes to the excitotoxic effects in an AD brain. The altered tryptophan levels consequently affect 5HT synthesis, which is a neurochemical hallmark in the etiology of depression. Neuroinflammation by upregulating IDO and consequently lowering tryptophan levels has therefore been linked with major depressive disorder in AD patients (Dobos et al. 2010).
11.1.3.2 Other Dementia Subtypes
Except for AD which is the most prevalent dementia syndrome (65 % approximately), AD with cerebrovascular disease (AD + CVD), vascular dementia (VAD), dementia with Lewy bodies (DLB), Parkinson’s disease dementia (PDD), and frontotemporal dementia (FTD) together roughly account for the other 35 % (Fig. 11.4) (Small et al. 1997).
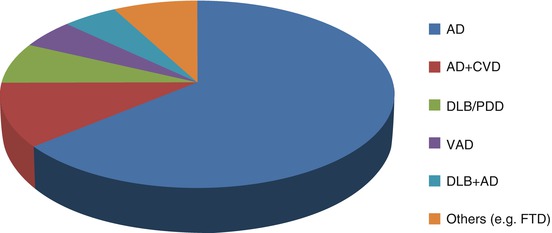
Fig. 11.4
Different etiological diagnoses of dementia. Alzheimer’s disease is the most prevalent dementia subtype (64 %), followed by Alzheimer’s disease + cerebrovascular disease (AD + CVD) (11 %), dementia with Lewy bodies (DLB)/Parkinson’s disease dementia (PDD) (7 %), vascular dementia (VAD) (5 %), dementia with Lewy bodies and Alzheimer’s disease (DLB + AD) (5 %), and finally other types of dementia (8 %), such as frontotemporal dementia (FTD) (Based upon Small et al. (1997))
Below, DLB and FTD are briefly described as they significantly differ from AD concerning their diagnostic criteria, pathogenesis, disease course, and behavioral profiles.
Dementia with Lewy Bodies (DLB)
Dementia with Lewy bodies (DLB) is the third most prevalent dementia subtype and is diagnosed according to McKeith et al. (2005). Comparable with AD, several core and supportive criteria need to be present in order to establish a clinically acceptable DLB diagnosis. The three core criteria are a fluctuating cognition, recurrent and well-described visual hallucinations, and clinical sings of parkinsonism (extrapyramidal symptoms (EPS): tremor, rigidity, and hypokinesia). The presence of only two core criteria is sufficient to diagnose “probable” DLB. Some other supportive criteria are a disturbed REM sleep behavior, a low dopamine transporter reuptake in the basal ganglia proven on SPECT or PET imaging, autonomic dysfunction, depression, and concurrent delusional ideation. Furthermore, DLB patients suffer from neuroleptic sensitivity which severely worsens EPS when classical neuroleptics (antipsychotic medication) are administered (McKeith et al. 2005). The difference between DLB and PDD is solely based upon the temporal sequence of appearance of the extrapyramidal symptoms: DLB should be diagnosed when dementia occurs before (at least 1 year in research studies) or concurrently with parkinsonism (if it is present). The term PDD should be used to describe dementia that occurs in the context of well-established Parkinson’s disease (PD) (Geser et al. 2005; McKeith et al. 2005).
The main pathological characteristic of DLB is the presence of cytoplasmatic aggregated inclusions of α-synucleins, generally known as “Lewy bodies” (Vladimir 2007). Synucleinopathies form a group of neurodegenerative disorders that share common pathologic proteinaceous lesions containing aggregated α-synuclein molecules which are deposited in vulnerable positions of neurons and glia (Goedert 1999;2001). Specifically in DLB, Lewy body aggregates precipitate in the substantia nigra (pars compacta) of the basal ganglia and also in the neocortex and hippocampus (McKeith et al. 2005). Only when a loss of dopaminergic neurons of 80 % or more in the substantia nigra is reached, EPS will set off. Several case studies demonstrated the occurrence of familial DLB cases (Gwinn-Hardy and Singleton 2002) and that Lewy bodies are commonly seen in familial cases of AD as well (Trembath et al. 2003). There are reports of triplications of the α-synuclein (SNCA) gene in DLB, PD, and PDD patients, whereas SNCA gene duplications only seem to be associated with motor PD, suggesting a possible gene dose effect (Singleton and Gwinn-Hardy 2004). However, SNCA gene multiplications were not found in most sporadic DLB cases (Johnson et al. 2004).
Frontotemporal Dementia (FTD)
A less frequent neurodegenerative disorder is frontotemporal dementia (FTD). Neary et al. (1998) established the diagnostic criteria of among others FTD, which forms one of the three diagnostic entities of “frontotemporal lobar degeneration (FTLD)” together with primary progressive aphasia and semantic dementia (SD). Typical for FTD patients is the very early disease onset compared to AD or DLB, namely, between the age of 45 and 70. At onset of the syndrome, there may typically be a neglect of personal hygiene, disinhibition, loss of insight and judgement, social neglect, and emotional disturbance (i.e., emotional bluntness, impaired control of emotions) in contrast to a comparatively spared memory and spatial abilities (core criteria). A subsequent cognitive impairment is inevitable although in the beginning amnesia remains surprisingly absent. FTD thus initially manifests itself by subtle changes in behavior and character (De Deyn et al. 2005; Neary et al. 1998). Some other typical behavioral characteristics are the expression of stereotypes and changes in sexual behavior, dietary hyperactivity, speech disturbances (echolalia, mutism, logorrhea), and restlessness. From a clinical point of view, FTD is also likely to be recognized and distinguished from AD solely due to this distinctive behavioral pattern (De Deyn et al. 2005).
Similarly as with AD, FTD can be subdivided into familial (±30 %) and sporadic (±70 %) variants. For familial FTD, a distinction must be made between tauopathies and non-tauopathies. Tauopathies are caused by a mutation in the microtubule-associated protein tau (MAPT) gene (Bancher et al. 1987; Sieben et al. 2012), whereas non-tauopathies can be etiologically defined by mutations in the progranulin (PGRN) (Cruts et al. 2006) and TAR DNA-binding protein 43 (TDP-43) gene (Arai et al. 2006; Neumann et al. 2006). Mutations in the MAPT gene cause cytoplasmatic tau to aggregate which leads to the formation of tangles and eventually to neuronal death, especially in frontotemporal cortical areas. Neuropathologically, this degenerative phenomenon is known as “Pick’s disease,” but supranuclear palsy and corticobasal degeneration are also classified as tauopathies (Keith 2008). On the other hand, mutations in both TDP-43 and PGRN genes (non-tauopathies) cause TDP-43 and PGRN aggregates, again leading to a consequent neuronal degradation (Sieben et al. 2012). Histopathologically, these aggregates are visible as tau-negative but ubiquitin (U)-positive inclusions so that non-tauopathies are generally categorized as FTLD-U. Noteworthy, the frontotemporal localization of tau and ubiquitin lesions in FTLD patients is pathophysiologically crucial to cause the frontal behavioral phenotype clarified above.
Recently, Gijselinck et al. (2012) identified a pathogenic GGGGCC repeat expansion in the C9orf72 promoter region on chromosome 9p21 in FTLD and amyotrophic lateral sclerosis (ALS) patients of a Flanders-Belgian cohort. FTLD and ALS are both clinically, pathologically, and genetically overlapping degenerative diseases. This genetic linkage and association study was performed in 337 FTLD, 23 FTLD-ALS, 141 ALS, and 859 control subjects. The GGGGCC repeat expansion showed to be highly penetrant, explaining all of the contribution of chromosome 9p21 to FTLD and ALS in this cohort. As for now, the function of the C9orf72 gene remains unknown, although it is highly conserved in all vertebrates. Further research of its function might eventually lead to a better insight into the common pathophysiological mechanisms of FTLD and ALS.
11.2 Behavioral and Psychological Signs and Symptoms of Dementia (BPSD)
Besides cognitive disturbances, dementia is characterized by numerous behavioral disturbances as well, categorized as Behavioral and Psychological Signs and Symptoms of Dementia (BPSD) (Finkel et al. 1996; Reisberg et al. 1987). BPSD are a heterogeneous group of behavioral, psychological, and psychiatric disturbances occurring in 50–80 % of dementia patients of any etiology (Finkel et al. 1996). These behavioral and psychological symptoms are generally classified into seven main subtypes: paranoid and delusional ideation, hallucinations, activity disturbances, aggressiveness, diurnal rhythm disturbances, affective disturbances, and anxieties/phobias (Reisberg et al. 1987). BPSD often lead to a greater amount of caregiver distress, diminished quality of life for both patient and caregiver, greater cognitive impairment (Weamer et al. 2009), premature institutionalization, frequent (re)hospitalizations, and increased secondary morbidity and mortality (Finkel 2000). Last but not least, BPSD also have a significant and increasing socioeconomic impact (Beeri et al. 2002) (Fig. 11.1).
From an etiological point of view, research has repeatedly suggested that there is a neurochemical basis underlying BPSD although its pathophysiological mechanisms are still not well understood (Engelborghs et al. 2008). Alterations in central noradrenergic (Engelborghs et al. 2008; Herrmann et al. 2004; Lanari et al. 2006; Matthews et al. 2002), serotonergic (Engelborghs et al. 2008; Garcia-Alloza et al. 2005; Lanctôt et al. 2001), and dopaminergic (Engelborghs et al. 2008; Lanari et al. 2006) neurotransmitter systems and associated receptors proved to play a critical role in BPSD manifestation, irrespective of the dementia subtype (Vermeiren et al. 2012). Particularly the balance between those different neurotransmitter systems seems to be of importance as it is conceivable, due to the neurochemical complexity and diversity of BPSD, that more than one neurotransmitter system contributes to a particular behavioral syndrome (Lanari et al. 2006). Studying neurotransmitter systems in isolation cannot fully explain changes in behavior, given that many neurotransmitter systems work in conjunction with each other. In spite of this difficulty, the neurochemical mechanisms underlying BPSD are proven to be both BPSD- and dementia-specific (Engelborghs et al. 2008; Vermeiren et al. 2012), so that dementia-specific neurochemical alterations might be found. There is also supportive evidence for amino acids playing a functional role in the neurochemical pathophysiology of BPSD (Engelborghs et al. 2003; Fekkes et al. 1998; Francis 2009; Garcia-Alloza et al. 2006), with, e.g., significantly high correlations between CSF taurine levels and depression in AD and CSF glutamate levels and agitation in FTD (Vermeiren et al. 2012).
Engelborghs et al. (2005) showed that different behavioral patterns can be observed depending on the dementia subtype, thereby further stressing that behavioral assessment itself may help in differentiating between different forms of dementia (Fig. 11.5).
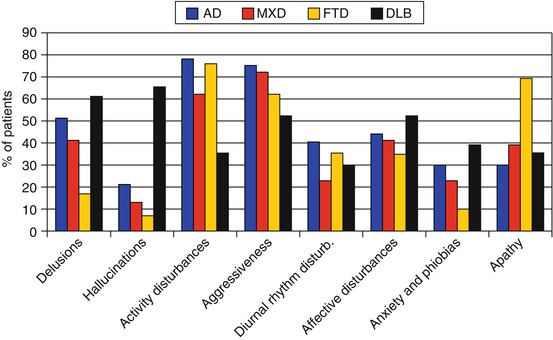
Fig. 11.5
Frequency of dementia-specific BPSD items. This figure shows that, e.g., apathy is much more frequent in FTD as compared to AD/MXD/DLB whereas delusions, hallucinations, and anxieties are less frequently present in FTD compared to DLB. Abbreviations: AD Alzheimer’s disease, BPSD Behavioral and Psychological Signs and Symptoms of Dementia, DLB dementia with Lewy bodies, FTD frontotemporal dementia, MXD mixed dementia (Based upon Engelborghs et al. (2005))
In 1996, Jost and Grossberg examined the frequency of BPSD in temporal relationship with the diagnostic progression of AD patients, as is demonstrated in Fig. 11.6. In contrast to the cognitive symptoms in AD which progressively worsen during its course, BPSD are different as some behavioral symptoms are severely present during the early disease stages (e.g., depression) although later on these symptoms might gradually diminish or even completely disappear, to be eventually replaced by other BPSD items (e.g., aggression).
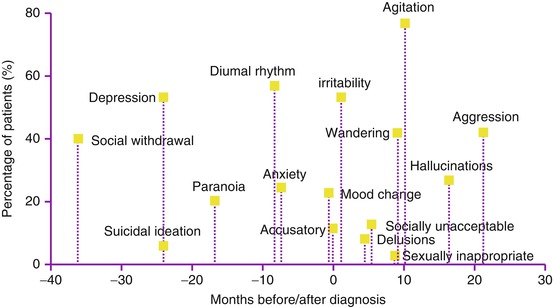
Fig. 11.6
Frequency of BPSD in temporal relationship with the progression of AD diagnosis. The evolution of Behavioral and Psychological Signs and Symptoms of Dementia (BPSD) in 100 autopsy-confirmed Alzheimer’s disease (AD) patients before and after their initially established diagnosis is shown above. Especially depression and diurnal rhythm disturbances seem to be significantly present roughly 25 and 9 months before AD diagnosis whereas aggression, agitation, and hallucinations are symptoms that are characteristically manifested approximately 2–3 years later (Based upon Jost and Grossberg (1996))
11.2.1 Delusional Ideation and Hallucinations: The Psychotic Syndrome
Approximately more than 40 % of dementia patients of any etiology and up to 73 % of AD patients suffer from delusional ideation during the disease course (Finkel 2001). The most prominent delusion according to Reisberg et al. (1987) is suspiciousness/paranoia, i.e., the conviction that people are stealing things from the patient. Other frequently occurring delusions are the “one’s house is not one’s home delusion” or the accusation of infidelity towards their spouse or caregiver. Delusions are frequently associated with verbal and physical aggression which in most cases leads to an untenable situation at home and premature institutionalization (Deutsch et al. 1991). Deutsch et al. (1991) suggest delusions to be risk factors in patients with probable AD who have moderate to severe cognitive impairment.
In patients with AD, psychosis occurs more frequently in women than in men. Some other predisposing factors besides gender for psychotic symptoms are age, severity of illness, and cognitive deterioration (Hirono et al. 1998). Weamer et al. (2009) found that the severity of cognitive impairment was a strong predictor of psychosis in AD patients up to 2 years prior to psychosis onset.
Hallucinations in dementia patients are less frequent than delusions, with a prevalence rate of 12 up to 49 % (Swearer 1994). Hallucinations as well as delusions are characteristic for specifically DLB patients, as is shown in Fig. 11.5 (Engelborghs et al. 2005). A hallucination is the patient’s strict conviction of a sensory perception in the absence of sensorial stimulation. Reisberg et al. (1987) made a distinction between visual, auditory, olfactory (smell), and haptic (touch) hallucinations. It is noticeable that hallucinations are more likely to occur in patients with more severe cognitive deterioration compared to patients with mild forms of dementia (Devenand et al. 1997). Moreover, hallucinations are less stressful for dementia patients than delusions so that pharmacological treatment is less mandatory (De Deyn 2004).
AD patients with psychosis have been reported to deteriorate twice as fast as patients without psychotic symptoms (Rosen and Zubenko 1991). Similarly, Scarmeas et al. (2005) studied whether the presence of delusions and hallucinations has predictive value for important outcomes in AD patients, such as cognitive and functional decline. Their results confirmed that the presence of delusions and hallucinations was associated with an increased risk for cognitive and functional decline, institutionalization, and even death.
It is noteworthy that psychosis of AD is a distinct syndrome that is markedly different from, e.g., schizophrenia in elderly patients. Numerous research groups have reported potentially relevant clinical, neuropsychological, neurochemical, neurobiological, and neuropathological differences between AD patients with and without psychosis (Jeste and Finkel 2000). In the past, there have been no specific criteria for diagnosing psychosis of AD as a distinct entity. Therefore, Jeste and Finkel have proposed several core criteria in 2000 in order to correctly diagnose the psychotic syndrome in AD. Characteristic symptoms are the presence of one (or more) visual/auditory hallucination(s) and/or delusion(s). Secondly, there has to be evidence from the patient’s history that these symptoms have not been continuously present prior to dementia onset. The symptoms also must have been present for at least 1 month or longer and have to cause some disruption in the patient’s functioning. Moreover, schizophrenia and related psychotic disorders as well as a delirium or other causes (e.g., substance-related) that might have initiated the psychosis need to be excluded. Finally, associated behavioral features such as agitation, negative symptoms, and/or depression might be present as well.
All criteria may also apply to a similar psychotic syndrome associated with other dementias such as DLB, VAD, and MXD.
11.2.2 Agitation and Aggression
Agitation includes inappropriate verbal, vocal, or motor behaviors that, in the opinion of an observer, do not result directly from the needs or confusion of the agitated individual (Cohen-Mansfield and Deutsch 1996). Approximately 80 % of dementia patients will suffer from agitation during the disease course. Agitation therefore is one of the most frequently (re)occurring BPSD (Allen and Burns 1995). In 2000, Lyketsos et al. reported the prevalence of agitation and other BPSD in 329 participants with dementia (the Cache County Study on Memory in Aging, Utah), of which 65 % had AD, and concluded that agitation and aggression were present in approximately 24 % of dementia patients. Given that the estimates were only considered over 1 month before behavioral assessments and due to the episodic course of this behavioral symptom, Lyketsos et al. (2000) mentioned that these prevalence numbers were an underestimation of the cumulative prevalence which may approach 70–80 %. Subsequently, the Cache County Study was resumed in 2003 (Steinberg et al. 2008) in which an incident sample of 408 dementia participants was behaviorally assessed during a 5-year follow-up period. At the end, 42 % of dementia participants developed agitation.
In general, agitation mostly occurs in the moderate stages of dementia and less in mild or severe dementia stages (Cohen-Mansfield et al. 1989; Lyketsos et al. 2000). Cohen-Mansfield et al. (1989) make a distinction between physically non-agitated behavior (e.g., restlessness, pacing, cognitive abulia, wandering, inappropriate (dis)robing) and verbally agitated behavior (e.g., negativism, complaining, repetitive sentences or questions, strange noises, unwarranted request for attention).
Aggression has a frequency between 20 and 30 % (Allen and Burns 1995) and can be divided into physically aggressive behavior (e.g., hitting, kicking, pushing, scratching, biting) and verbally aggressive behavior (e.g., screaming, cursing) (De Deyn 2004). In general, physically aggressive behavior is more common in male dementia patients compared to females (Cohen-Mansfield and Deutsch 1996). Furthermore, aggression in dementia patients is associated with depression according to Lyketsos et al. (1999).
11.2.3 Diurnal Rhythm Disturbances
Sleep disturbances can be subdivided into difficulties falling asleep, multiple awakenings during sleep, early morning awakenings, or a completely inversed sleep-wake pattern (Prinz et al. 1982). Insomnia in dementia also seems to be the most prominent reason for an eventual institutionalization according to Harper et al. (2001). One specific diurnal rhythm disturbance is sundowning, a situation in which patients are relatively calm during the day but as evening falls show an exacerbation of behavioral symptoms, such as pacing, wandering, and repetitive, purposeless activities (cognitive abulia) (Little et al. 1995).
11.2.4 Depression
In AD, depression has a prevalence of 20 (Castilla-Puentes and Habeych 2010) up to 50 % (Starkstein et al. 2005). As shown in Fig. 11.6, depression is mostly present in mild to moderate AD or even 2 years before the established AD diagnosis (Alexopoulos et al. 1988; Jost and Grossberg 1996). A major depressive episode in dementia is characterized by mood-related signs (anxiety, lack of reactivity to pleasant events, irritability), behavioral symptoms (agitation, retardation (slow movements and speech), loss of interest, physical complaints), physical signs (appetite and weight loss, lack of energy), sleep rhythm disturbances, and ideational disturbances (pessimism, suicidal wishes, poor self-esteem) (Alexopoulos et al. 1988). Besides the behavioral aspects, depression is also characterized by deficits in verbal and visual memory, concentration, and executive functioning (Sierksma et al. 2010). Several research groups have even suggested that depression in general might be a prodrome (i.e., a premonitory symptom indicating the onset of a disease; risk factor) of developing AD (Caraci et al. 2010; Korczyn and Halperin 2009), given the fact that the pathophysiological properties of depression and some etiological hallmarks of AD are related (e.g., increased neuroinflammation, monoaminergic deficiency, increased synaptic neurodegeneration, and altered neurotrophic factors) (Sierksma et al. 2010). Depressed dementia patients also have a higher mortality rate compared to their nondepressed counterparts (Rovner et al. 1991).
11.2.5 Activity Disturbances
According to Reisberg et al. (1987), activity disturbances form a separate entity in the behavioral phenomenology of AD patients among others. Approximately 80 % of AD patients suffer from activity disturbances (Engelborghs et al. 2005), which can be best described as a form of physical agitation. Some examples are wandering, purposeless activities (e.g., cognitive abulia, such as repetitive (dis)robing, pacing), and inappropriate activities (inappropriate physical sexual advances, hiding objects, hoarding) (Reisberg et al. 1987). In some cases, activity disturbances are severe enough to require restraint or even result in abrasions (e.g., pacing) or physical harm. Besides AD, FTD patients characteristically suffer from certain types of activity disturbances as well, mainly stereotype movements (e.g., tapping, hand clapping, patting, hand rubbing, wandering a fixed route) and general restlessness (aimless wandering, pacing, fidgeting, inability to sit still) (De Deyn et al. 2005).
11.2.6 Anxieties and Phobias
Although less frequent, anxiety is a psychological symptom in dementia patients which is present in different variants (De Deyn 2004). The anxiety or fear of being left alone as well as the Godot syndrome are two frequent types of anxiety in AD patients (Reisberg et al. 1987). In case of Godot syndrome, patients repeatedly and constantly ask questions concerning a completely normal but approaching event such as a meeting with the family doctor (Reisberg et al. 1986). This term was firstly described in the late 80s by Reisberg et al. (1986) and is an extreme form of anxiety in dementia patients and sometimes requires the patient to be accompanied at all times. On the other hand, pacing, stereotype behavior, and restlessness might be physical reflections of a rooted anxiety residing within the patient. A phobia is an anxiety disorder which is disproportional to the actual danger, often being irrational. Examples are fear of traveling, bathing, darkness, and overcrowded places (De Deyn 2004).
11.2.7 Apathy
In the context of dementia, apathy has been recently defined as a disorder of diminished motivation that persists over time for at least 4 weeks with an additional reduced goal-directed behavior, cognitive activity, and emotions (Robert et al. 2009). These relatively new criteria have been established due to the overlap between apathy and depression among others. Apathy is a common behavioral disorder not only in AD but also in PD, FTD, and stroke (Levy et al. 1998). Results from the European Alzheimer’s Disease Consortium study in 2007 showed that apathy is the most prominent and persistent neuropsychiatric syndrome in dementia as it occurred in 65 % of the total 2,354 AD patients (Aalten et al. 2007). Additionally, it is also present during all stages of the disease (Lyketsos et al. 2011; Robert et al. 2009), and there is a growing body of evidence that it might be indicative of a pre-dementia state (Ready et al. 2003; Robert et al. 2009).
11.3 Behavioral Assessment Scales
In order to evaluate this large group of behavioral and neuropsychiatric symptoms in dementia patients, different behavioral assessment scales have been developed throughout the years. The most common are described below, i.e., Middelheim Frontality Score (MFS), Behavioral Pathology in Alzheimer’s Disease Rating Scale (Behave-AD), Cohen-Mansfield Agitation Inventory (CMAI), Geriatric Depression Scale (GDS), Cornell Scale for Depression in Dementia (CSDD), and Neuropsychiatric Inventory (NPI). All these scales are very useful assessment tools to identify the behavioral profile of dementia patients or even to distinguish between different types of dementia (De Deyn et al. 2005). The efficacy of novel psychotropic medication in the treatment of BPSD can also be demonstrated by the use of these well-validated and drug-sensitive behavioral scales mentioned above, such as Behave-AD, CMAI, and NPI (De Deyn and Wirshing 2001). Moreover, these behavioral assessment scales are widely used to study the neuroanatomical and pathophysiological etiology of different behavioral phenotypes in dementia in combination with neuroimaging data.
11.3.1 Middelheim Frontality Score (MFS)
The Middelheim Frontality Score (MFS) is a clinical and behavioral assessment tool which measures frontal lobe features and secondly, in contrast to classical behavioral scales, reliably discriminates FTD from AD patients (De Deyn et al. 2005). The MFS is rated by a clinician and is obtained by summating the scores in a standardized fashion on ten different items. Each item is scored either zero (absent) or one (present), yielding a total maximal score of 10. Information is obtained through an interview of the patient and her/his professional and/or main caregiver, clinical files, and behavioral observation. The ten items are (item 1) initially comparatively spared memory and spatial abilities that reflect the neurobehavioral onset of the disease; frequently occurring personality and behavioral changes like (item 2) loss of insight and judgement; (item 3) disinhibition; (item 4) dietary hyperactivity (referring to overeating); (item 5) changes in sexual behavior (hypersexuality as well as the more frequently occurring hyposexuality); (item 6) stereotyped behavior (encompasses all kinds of stereotyped behavior, both simple repetitive behaviors (can also be oral) and complex behavioral routines such as wandering); (item 7) impaired control of emotions, euphoria, or emotional bluntness; (item 8) aspontaneity; (item 9) speech disturbances such as stereotyped phrases, logorrhoea, echolalia, and mutism; and finally, (item 10) restlessness. Although the NPI is able to correctly classify 77 % of AD and FTD patients (Levy et al. 1996), the frequently used Behave-AD and CMAI lack sensitivity for FTD as they have been specifically developed for AD patients. The Behave-AD even underestimates BPSD in FTD patients as was shown by Engelborghs et al. (2004): 28 FTD patients had significantly lower Behave-AD total scores compared to 152 AD patients, whereas the Behave-AD global scores (reflecting caregiver burden) were not different between both patient groups. Moreover, Pickut et al. (1997) previously showed that the total MFS scores correlated with severity of bifrontal hyperperfusion on SPECT in FTD.
The discriminatory cutoff score of the MFS is set at a total score of 5 as, respectively, 85.9 and 76.6 % of clinically diagnosed FTD and AD patients were correctly classified (De Deyn et al. 2005).
11.3.2 Behavioral Pathology in Alzheimer’s Disease Rating Scale (Behave-AD)
In 1987, the Behavioral Pathology in Alzheimer’s Disease Rating Scale (Behave-AD) was developed to correctly assess and categorize frequently occurring behavioral symptoms of AD patients (Reisberg et al. 1987). The first part of the Behave-AD comprises 25 items of which each item can be rated from zero (absent) to three (severely present, with emotional and physical component, possibly requiring restricting) with a total maximum score of 75. The second part is the Behave-AD global score which assesses caregiver burden: 0 (not at all troubling to the caregiver or dangerous to the patient), 1 (mildly troubling to the caregiver or dangerous to the patient), 2 (moderately troubling to the caregiver or dangerous to the patient), and 3 (severely troubling to the caregiver or dangerous to the patient). The first 25 items are categorized into 7 behavioral clusters: cluster A (paranoid and delusional ideation, items 1–7), cluster B (hallucinations, items 8–12), cluster C (activity disturbances, items 13–15), cluster D (agitation and aggression, items 16–18), cluster E (diurnal rhythm disturbances, item 19), cluster F (affective disturbances, items 20–21), and cluster G (anxieties and phobias, items 22–25).
The Behave-AD is a very detailed and relatively simple scale which allows an assessment within a short amount of time (De Deyn 2004). Several studies (Sclan et al. 1996; Patterson et al. 1990) showed that the reliability of the Behave-AD is comparable with those of several widely used cognitive assessment scales, such as the Mini-Mental State Examination (MMSE) (Folstein et al. 1975). However, one disadvantage of the Behave-AD is its specificity for and usage in exclusively AD patients. Furthermore, only the intensity of the 25 BPSD items is rated (scores 0–3) and not the frequency (De Deyn 2004).
11.3.3 Cohen-Mansfield Agitation Inventory (CMAI)
The Cohen-Mansfield Agitation Inventory (CMAI) was originally designed for the staff of nursing homes to rate the frequency of agitation and related behaviors in the elderly with cognitive deterioration. This scale assesses 29 types of agitated behavior which are subdivided into 3 main categories: items 1–10 comprise “aggressive behavior,” items 11–21 consist of “physically nonaggressive behavior,” and finally items 22–29 are clustered into the category “verbally agitated behavior.” Each item is scored depending on its frequency, i.e., from 1 (never) to 7 (several times an hour) (Cohen-Mansfield et al. 1989).
11.3.4 Geriatric Depression Scale (GDS)
The Geriatric Depression Scale (GDS) is the oldest scale so far and was designed to estimate depression in non-demented elderly (Yesavage et al. 1983). It takes little or no experience for the investigator to use this scale which consists of 30 questions that are related to depression in the elderly. Each question should be answered with a simple “yes” or “no.” A score of 12 or more is indicative of a “light” depression whereas 18 or more point to moderate depression. Debruyne et al. (2009), using the CSDD as the golden standard, concluded that the GDS-30, is not a reliable screening tool when assessing depressive symptoms in dementia patients but only in patients with mild cognitive impairment (MCI) and non-demented elderly.
11.3.5 Cornell Scale for Depression in Dementia (CSDD)
The Cornell Scale for Depression in Dementia (CSDD) dates from 1988 and is a very useful assessment tool to diagnose depression in dementia (Alexopoulos et al. 1988). The scale is a 19-item clinician-administered instrument that uses information from interviews with both the patient and nursing staff members, a method suitable for dementia patients. Each item is scored based on a three-point scale, i.e., 0 (absent), 1 (mild or intermittent), and 2 (severely present). If it is impossible to rate one of the items, a score remains absent (A: unable to evaluate). All 19 items are subdivided into 5 main categories:
A.
Mood-related signs (anxiety, sadness, lack of reactivity to pleasant events, irritability)
B.
Behavioral disturbances (agitation, retardation (slow movements and speech), multiple physical complaints, loss of interest)
C.
Physical signs (appetite loss, weight loss, lack of energy)
D.
Cyclic functions (diurnal variation of mood, diurnal rhythm disturbances)
E.
Ideational disturbances (suicidal ideation, poor self-esteem, pessimism, mood-congruent delusions).
A score of 8 or more is suggestive for the presence of depression (Burns et al. 2004).
11.3.6 Neuropsychiatric Inventory (NPI)
The Neuropsychiatric Inventory (NPI) evaluates 12 types of behavioral disturbances that are dementia-specific, i.e., delusions, hallucinations, agitation/aggression, depression/dysphoria, anxiety, euphoria, apathy/indifference, disinhibition, irritability/lability, repetitive purposeless behavior, insomnia/diurnal rhythm disturbances, and appetite or a change in dietary activity (Cummings et al. 1994). The severity and the frequency of these symptoms are rated by a series of questions which are intended for the main caregiver of the patient. The severity score is based on a three-point scale ranging from 1 (mild) to 3 (severe), and the frequency score can vary between 1 (occasionally, less than once a week) and 4 (very frequent, multiple times a day). The scores of each of these 12 behavioral symptoms need to be summed up to obtain a total NPI score. Besides the severity and frequency scores, the level of caregiver distress (emotional burden) of each of the 12 behavioral symptoms requires rating as well. In this case, a scale ranging from 0 (no distress) to 5 (severe and extreme distress) is provided (Kaufer et al. 1998). The total score of “caregiver distress” is yielded by summing up the 12 individual distress subscores.
Because the NPI consists of a gross variety of behavioral symptoms, it is a useful instrument to discriminate between different types of dementia as well as to evaluate the behavioral outcome due to pharmacological interventions (De Deyn 2004; De Deyn and Wirshing 2001). In 2001, a shortened version of the NPI, namely, NPI-Q (questionnaire), was developed by Kaufer et al. (2000) which facilitates its daily use in a clinical setting. Several other forms of the NPI have also been proposed depending on the informant, such as clinicians (NPI-C) (de Medeiros et al. 2010), or the institutional setting, such as nursing homes (NPI-NH) (Wood et al. 2000).
11.4 PET in the Differential Diagnosis of Dementia
Neuroimaging has played an important role in the study and differential diagnosis of dementia over the last 40 years. More recently, positron emission tomography (PET) studies of cerebral metabolism with 18F-fluorodeoxyglucose (FDG) and amyloid tracers such as the Pittsburgh Compound-B (PiB) have provided invaluable information regarding specific AD-like brain changes (Johnson et al. 2012). Even in prodromal and presymptomatic states, PET imaging has emerged as a robust biomarker of neurodegeneration in individuals who were later found to progress to AD (de Leon et al. 2001; Bateman et al. 2012). Bateman et al. (2012), for example, detected early Aβ-deposition in the precuneus of 128 autosomal dominant AD patients measured by PET-PiB nearly 15 years before expected symptom onset, indicating PET imaging to be an essential and reliable imaging tool not only in the differential diagnosis between AD and non-AD but even in asymptomatic states.
Some of the most important PET radioligands and compounds which are widely used in the differential diagnosis of dementia are described below.
11.4.1 Radioligands and Compounds
Brain FDG-PET primarily indicates synaptic activity. Because the brain relies almost exclusively on glucose as its main energy resource, the glucose analog FDG is suitable as an indicator of brain metabolism and, when labeled with fluorine-18 (18F) (half-life 110 min), is detected with PET. Especially, the glutamatergic synaptic signaling is responsible for the maintenance of intrinsic, resting (task-independent) activity of the cerebral cortex which, most of the time, is the brains main task (Johnson et al. 2012; Sibson et al. 1997). Therefore, [18F]-FDG-PET is widely accepted to be a valid biomarker of the overall brain metabolism to which ionic gradient maintenance for synaptic activity is the most principal contributor (Schwartz et al. 1979; Magistretti 2006). The characteristic pattern found in AD generally is a hypometabolism of the temporoparietal cortex (Herholz et al. 2002; Ferreira and Busatto 2011) and specific limbic and association areas, such as the precuneus, posterior cingulate gyri, inferior parietal lobes, and posterolateral portions of the temporal lobe as well as the hippocampus and medial temporal cortices (Foster et al. 1983; Minoshima et al. 1997; Reiman et al. 2005). An asymmetry between both hemispheres is commonly seen in the early stages of AD whereas in a more advanced stage of the disease, usually the prefrontal association areas become affected (Johnson et al. 2012).
Recently, a meta-analysis showed that hypometabolism of the inferior parietal lobes and precuneus are the most striking neurological findings on FDG-PET imaging in AD patients compared to non-demented elderly (Schroeter et al. 2009). Moreover, longitudinal neurofunctional imaging studies have demonstrated hypometabolism in the parietal lobe of MCI converters in comparison with those who did not convert to AD (Schroeter et al. 2009). In conclusion, FDG-PET can be useful in cases of diagnostic uncertainty and has even shown to be valuable in distinguishing AD from FTD (Foster et al. 2007). However, it is advisable to always combine FDG-PET findings with imaging data of other neuroimaging techniques as FDG-PEt alone does not allow an adequate evaluation of the brain structure (Waldemar et al. 2007).
The pathological hallmark of the AD brain is the extracellular deposition of Aβ-plaques. Consequently, a second strategy to visualize AD pathology is not based on glucose metabolism, but on a synthesized derivate which in vivo binds Aβ, such as the N-methyl[11C]2-(4′methylaminophenyl)-6-hydroxybenzothiazole, also known as Pittsburgh Compound-B (PiB) (Mathis et al. 2002), [125I]-6 (Wang et al. 2002), and [3H]-BTA-1 (Klunk et al. 2003). All compounds have binding properties for Aβ in the nanomolar range and are based on thioflavin, a well-known chemical dye that stains a wide range of amyloid pathologies (Suhara et al. 2008). PET studies using PiB labeled with carbon 11 (11C) showed that amyloid deposition already occurs years before the clinical diagnosis of dementia (Chetelat et al. 2010), is related to cortical atrophy rate as well as cognitive decline (Braskie et al. 2010), and is more present in MCI converters compared to non-converters (Forsberg et al. 2008). One concern however is the short half-life of PiB labeled with 11C, which renders its use in some diagnostic clinical settings more difficult. Consequently the interest has raised to develop an amyloid-sensitive, radioactive-labeled PiB with longer half-life, such as PiB labeled with 18F (Wong et al. 2010). A very promising 18F-labeled amyloid imaging tracer which has recently been FDA-approved (April 6, 2012) is 18F-florbetapir (18F-AV-45) (Choi et al. 2009). Recent studies have compared the diagnostic utility of [18F]-florbetapir-PET compared to [11C]-PiB-PET (Wolk et al. 2012) and the commonly used [18F]-FDG-PET (Newberg et al. 2012), concluding that [18F]-florbetapir-PET produced comparable results in discriminating AD patients from cognitively normal adults. Doraiswamy et al. (2012) even proved that [18F]-florbetapir-PET may help in identifying individuals who are at increased risk for progressive cognitive decline. The same goes for florbetaben (BAY 94–9172), another valuable 18F-PET marker for Aβ imaging which is currently in phase III clinical development with a sensitivity and specificity of 80 and 91 % in an AD versus control comparison (Barthel and Sabri 2011). Last in the series of 18F-labeled amyloid PET imaging tracers is 18F-flutemetamol (phase III trial). Recent work demonstrated similar findings of 18F-flutemetamol in probable AD and MCI patients relatively to healthy controls with a similar performance as 11C-PiB within the same subjects (Vandenberghe et al. 2010). Additionally, Wolk et al. (2011) demonstrated a high correspondence between immunohistochemical estimates of Aβ levels in brain tissue of 7 AD patients who underwent previous biopsy and in vivo quantitative measures of 18F-flutemetamol uptake at the location contralateral to the biopsy site (i.e., right frontal), supporting its sensitivity to detect Aβ and its use in the study and early detection of AD.
Amyloid in vivo imaging is a very promising approach but is currently restricted to specialized centers around the world, although in the future it is likely that amyloid imaging techniques will be routinely used in the clinical evaluation of AD patients (Ferreira and Busatto 2011).
Besides amyloid deposits, intracellular NFT consisting of tau protein is a pathological feature of AD as well. The development of PET probes for in vivo imaging of NFT is presently an active research field (Ono and Saji 2012). The first 18F-labeled compound that was synthesized in order to bind NFT was [18F]-2-(1-(2-(N-(2-fluoroethyl)-N-methylamino)naphthalene-6-yl)ethylidene)malononitrile, abbreviated as FDDNP (Agdeppa et al. 2001; Barrio et al. 1999). Unfortunately, FDDNP does not exclusively bind NFT but also Aβ plaques. So, for the moment, no existing PET imaging agents allow an exclusive in vivo evaluation of tau pathology in AD brains (Ono and Saji 2012).
Finally, one last approach to visualize AD pathology using PET as an imaging tool is the in vivo mapping of altered neurochemical processes which are typical in the AD brain, such as cholinergic denervation (Van Dam and De Deyn 2006). One example is N-[11C]-methylpiperidin-4-yl propionate, known as [11C]-PMP (Kuhl et al. 1999). This novel radiopharmaceutical is used in PET imaging to determine the activity of the acetylcholinergic neurotransmitter system by acting as a substrate for acetylcholinesterase. Besides the cholinergic neurotransmission, PET imaging with radioligands that are involved with several other neurotransmitter systems or receptors, such as substrates for dopamine (DA) or serotonin (5HT) signaling, has provided important insights into several neurodegenerative disorders (Bohnen and Frey 2007) and has even helped in distinguishing AD from DLB and PD (Tatsch 2008).
Please visit http://www.clinicaltrials.gov/ to see ongoing clinical trials concerning the development of novel PET probes related to amyloid and tau imaging or other neurodegenerative-specific disease markers in the differential diagnosis of dementia.
11.5 PET Imaging in Neuropsychiatric Disturbances of Dementia
11.5.1 Alzheimer’s Disease
11.5.1.1 Depression and Apathy
Loss of neurons in the serotonergic raphe nuclei and dysfunction of its nerve terminals in the neocortex have been reported in AD (Mann and Yates 1983; Palmer et al. 1987). Many lines of evidence suggest this serotonin (5HT) deficiency theory to be strongly related with mood disorders in dementia patients and non-dementing elderly (Sierksma et al. 2010). In vivo imaging studies that used PET have so far focused on 5HT receptors in the limbic brain regions associated with cognitive impairment in AD (Kepe et al. 2006; Meltzer et al. 1998). Ouchi et al. (2009) more recently used a set of 2 different biomarkers in mild- to moderate-stage AD patients with and without depression to investigate the levels of presynaptic serotonergic function and cortical neuronal activity using PET with [11C]-DASB ([11C]-3-amino-4-(2-dimethylaminomethylphenylsulfanyl)-benzonitrile), a specific 5HT-transporter marker, and the more common [18F]-FDG-PET. Because the 5HT transporter is located on presynaptic 5HT terminals and regulates 5HT signaling, levels of [11C]-DASB binding in these regions thus reflect the activity of presynaptic 5HT neurons in the dorsal raphe nuclei. Thomas et al. (2006) previously found a marked reduction in the binding of 5HT-transporter levels in the prefrontal cortex of AD patients (n = 14) compared to control subjects (n = 10) and non-demented depressed subjects (n = 8), but not between depressed (n = 9) and nondepressed (n = 5) AD patients. Contrastingly, Ouchi et al. (2009) observed a negative correlation between [11C]-DASB binding potential levels in the subcortical serotonergic projection region (striatum) and GDS scores (n = 15) (Spearman Rank Order correlation, P < 0.01) as well as significantly lower [11C]-DASB binding potential levels in AD patients, irrespective of depression, compared to healthy controls (n = 10) in putamen, thalamus, and midbrain (P < 0.05). Consequently, Ouchi et al. (2009) suggested that a certain degree of presynaptic 5HT function in the subcortical 5HT-projection region is compromised in AD patients even before the development of depression. Secondly, statistical parametric mapping (SPM) correlation analysis showed that glucose metabolism in the right dorsolateral prefrontal cortex was positively associated with the levels of striatal [11C]-DASB binding, suggesting that right dorsolateral prefrontal dysfunction in parallel with 5HT inactivation is also implicated in the progression of emotional and cognitive deterioration in AD.
Related posts:
 SPECT and PET in Late-Life Depression
PET and SPECT Findings in Patients with Hallucinations
Neuroimaging Studies of Psychopathy
PET and SPECT Studies in the Chronic Fatigue Syndrome/Myalgic Encephalomyelitis
SPECT and PET in Late-Life Depression
PET and SPECT Findings in Patients with Hallucinations
Neuroimaging Studies of Psychopathy
PET and SPECT Studies in the Chronic Fatigue Syndrome/Myalgic Encephalomyelitis
 Dissociative Identity Disorder and Fantasy Proneness: A Positron Emission Tomography Study of Authentic and Enacted Dissociative Identity States
Dissociative Identity Disorder and Fantasy Proneness: A Positron Emission Tomography Study of Authentic and Enacted Dissociative Identity States
 Impulsivity Imaging
Impulsivity Imaging
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


