 CAUTION
CAUTIONSpongiform disorders
Creutzfeldt–Jakob disease is a rapidly progressive prion disorder of the CNS. Most patients have a sporadic form, although cases may be hereditary (5–15% of cases), iatrogenic (rare, occurring by transmission from infected corneal transplants, pooled cadaveric pituitary hormones, dural grafts, and electroencephalogram depth electrodes), and transmitted from cows infected with bovine spongiform encephalopathy (variant-CJD; younger onset-age [20s], and frequent psychiatric and sensory symptoms early on). For the sporadic form, average onset-age is 65 years, and sexes are affected equally. Clinical presentations vary, but the most frequent are changes in behavior and cognition, often followed by abnormalities in vision. Dementia and muteness usually ensue quickly, and myoclonus is prominent. Cerebellar ataxia (Oppenheimer variant) or visuospatial difficulties (Heidenhain variant) may predominate initially. The diagnosis rests on typical clinical findings, magnetic resonance imaging (MRI) abnormalities, and the exclusion of alternative causes. Routine CSF tests are normal; the diagnostic utility of 14-3-3 protein, S100B, and neuron-specific enolase (NSE) testing on CSF is controversial, with concerns regarding specificity of these tests. Data from the Case Western Reserve autopsy cohort suggest that 50% of patients with a potentially treatable disorder, previously diagnosed as CJD, had an elevated 14-3-3 protein.
Electroencephalogram (EEG) findings include an evolving pattern of diffuse slowing to high-voltage triphasic sharp wave (usually 1–2 Hz) complexes, that give the appearance of periodicity (pseudoperiodic). The latter EEG findings, though characteristic, typically appear during late stages of the disease.
Recent data suggest that cortical ribbon and deep lenticular nuclear diffusion-weighted (most striking) and fluid-attenuated inversion recovery (FLAIR) imaging abnormalities are the most sensitive and specific findings for CJD.
Rare spongiform disorders include Gerstmann–Straussler–Scheinker syndrome (autosomal dominant inheritance, chronic course with prominent corticospinal tract and extrapyramidal signs and relatively mild dementia) and fatal familial insomnia (autosomal dominant inheritance, rapid course with intractable insomnia and dementia).
Other rapidly progressive neurodegenerative dementias
At autopsy of patients with RPD, pathological evidence of neurodegenerative disorders other than prion disease is frequently detected. At the UCSF RPD unit, neurodegenerative disease (non-prion related) was responsible for 14.6% of all patients referred for suspected CJD and represented 39% of the non-prion cases. The Mayo Clinic series of 22 patients diagnosed with RPD showed that 23% of the cases consisted of frontotemporal dementia with motor neuron disease, 18% had either CBD or PSP, 14% had DLBD, and 9% had AD. In the Case Western Reserve autopsy cohort, AD accounted for over half of patients with a rapidly progressive incurable dementia diagnosed in life as CJD; other diagnoses in the non-prion neurodegenerative group were (in order of commonality) neurodegenerative disorder not otherwise specified, FTLD, DLBD, tauopathy not otherwise specified, PSP, CBD, and Huntington’s disease (HD).
Diagnostic clues useful for neurodegenerative disorders such as AD, DLBD, PSP, and CBD are usually unreliable when the clinical course is rapidly progressive. Illness duration beyond 12 months may be indicative of a neurodegenerative disorder other than CJD. In one series of six patients with RPD and autopsy diagnosis of DLBD, half presented with acute confusion to an emergency room. In the Mayo Clinic RPD series, patients with DLBD were distinguishable from other neurodegenerative causes on the basis of a transient postoperative or illness-related encephalopathy having occurred 2 years prior to the dementia onset. In the same series, parkinsonism, psychosis, rapidly fluctuating cognition (all typical of DLBD), and motor neuron disease (classically encountered in FTLD) were seen in patients with AD and CJD, and myoclonus was observed exclusively in CJD patients. Contrary to this, another series of autopsy-proven rapidly progressive DLBD demonstrated that half had myoclonus.
Indeed, multifocal signs, motor signs, psychosis, apathy, apraxia, and seizures have all been reported as signs predicting rapid progression in patients with AD in general. Other factors that have been reported to be associated with a rapidly progressive course in AD (all controversial) include co-morbid cardiovascular disease, diabetes mellitus, high educational level, low educational level, and severe cognitive impairment at disease onset. CSF measures associated with rapid cognitive decline in AD include high total tau or phosphorylated tau, low beta-amyloid Abeta1-42, and a high ratio of total tau to Abeta1-42.
Atypical presentations of other heredodegenerative disorders
Carriers of premutations of the FMR1 gene typically present with ataxia and intention tremor (the fragile X-associated tremor/ataxia syndrome). A syndrome dominated by RPD (executive dysfunction, apathetic behavior in addition to mild ataxia) has been described in two patients.
Psychiatric and cognitive changes may be initial manifestations of HD preceding motor signs by many years. The clinical presentation to a neurologist may be that of personality change, mood disturbance, and altered work performance. Although usually indolent in onset, physical or psychological stress may lead to fairly acute unmasking of clinical symptoms or signs. One patient with a RPD accompanied by generalized myoclonus, generalized tonic-clonic seizures, and ataxia was reported to have Unverricht–Lundborg disease, a form of progressive myoclonic epilepsy.
Autoimmune causes of rapidly progressive dementia
Autoimmune diseases may involve cerebral structures crucial for memory, cognition, and behavior. Careful consideration of autoimmune disorders presenting as RPD is important, since they are potentially amenable to immunotherapy if treated early.
Clinical terminology varies and includes autoimmune encephalitis, autoimmune dementia, limbic encephalitis (LE), paraneoplastic encephalopathy, and steroid-responsive encephalopathy with autoimmune thyroiditis (SREAT, aka Hashimoto encephalopathy). Over the last decade, identification of novel neural autoantibodies targeting proteins involved in synaptic transmission and plasticity has led to heightened awareness of autoimmune causes of cognitive impairment. Since many neural autoantibodies have only been identified recently, the phenotypical range of their associated neurological disorders continues to broaden.
In one series, young-onset dementia was attributed to autoimmune and inflammatory causes in 20% of patients. All ages can be affected; the largest study of voltage-gated potassium channel complex (VGKC) antibody seropositive patients reported a median onset-age of 65 years. Clinical presentations are heterogeneous and frequently multifocal.
Clinical manifestations
Patients with autoimmune neurological disorders may present acutely or subacutely. LE, a well-recognized autoimmune neurological disorder, is characterized by subacute short-term memory loss, neuropsychiatric disturbances (irritability, depression, hallucinations, personality change), and temporal lobe seizures.
In cases atypical for LE, an autoimmune cause may be overlooked. Other cognitive domains that may be affected in autoimmune encephalitis include learning, abstract reasoning, executive function, gnosis, and praxis. Attention deficits and impaired consciousness (delirium) are also commonly encountered in autoimmune cognitive decline, but their absence should not preclude the diagnosis of an autoimmune neurological disorder. The course of autoimmune dementia may fluctuate.
As accompanying neurological and psychiatric features are often encountered, history of behavioral changes, psychiatric symptoms (such as hallucination and mood changes), insomnia, movement disorders, seizures, and neuropathy should be obtained. In some, the multifocal neurological involvement may emerge only with temporal progression of the autoimmune disease. Personal or family history of autoimmunity and cancer are risk factors for autoimmune dementia, and their presence is helpful to raise further suspicion of an autoimmune etiology.
The clinical evaluation of cognitive function, using a cognitive test such as the Mini-Mental State Exam (MMSE) or Kokmen Short Test of Mental Status, should demonstrate impairments in one or more domains of cognition. Brainstem signs, myoclonus, ataxia, myelopathy, and peripheral somatic and autonomic neuropathy may be present. These features, indicative of a multifocal neurological involvement, may further trigger consideration of autoimmunity. A thorough clinical evaluation to search for non-neurological autoimmunity (alopecia, thyroid disease, vitiligo, systemic lupus erythematosus) should be performed.
Etiology and pathogenesis
When detected, neural-specific autoantibodies may serve as markers of the immune-mediated process. Neurological involvement may also be seen in non-organ specific autoimmune disorders, such as systemic lupus erythematosus and Sjogren syndrome; neural autoantibodies may not be detected in this setting.
Neural autoantibodies associated with autoimmune dementias
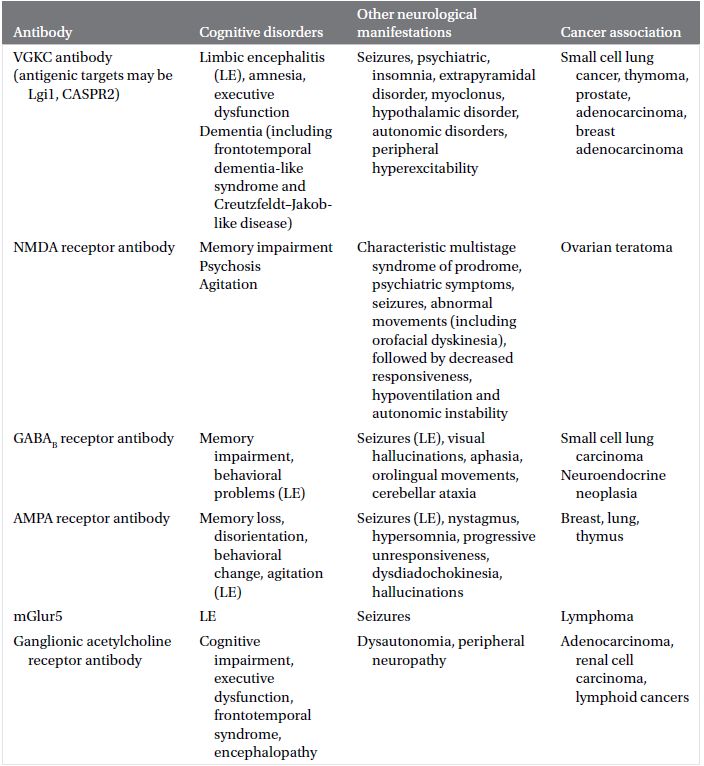
Neural autoantibodies (Plate 2.1) may be broadly classified under two categories, based on the location of antigenic targets: antibodies that target intracellular antigens (Table 2.1) and antibodies that target neural cell surface proteins or receptors (Table 2.2).
 SCIENCE REVISITED
SCIENCE REVISITED SCIENCE REVISITED
SCIENCE REVISITEDVGKC antibodies
Early associations of VGKC autoantibodies included peripheral nerve hyperexcitability (Isaacs syndrome, acquired neuromyotonia), Morvan syndrome, and LE. The cancer association of VGKC antibodies is relatively low in frequency (20–30%) and diverse in the range of tumors seen.
Cognitive impairment is the most common neurological manifestation, and these patients may present with a syndrome of RPD. Cognitive and neuropsychiatric manifestations include executive dysfunction, disorientation, agnosia, memory impairment, behavioral and personality changes (irritability, apathy, lethargy, aggression), depression, anxiety, hallucinations, and impaired attention. Seizures may be temporal or extratemporal in origin. Other multifocal neurological manifestations may occur. A hallmark feature described in these patients is the syndrome of inappropriate antidiuretic hormone secretion (SIADH) that is secondary to hypothalamic involvement.
Plate 2.1 Distribution of autoantibody immunoreactivity for ANNA-1 and NMDA receptor antibody in mouse neural tissues. ANNA-1 binds primarily to neuronal nuclei of cerebellar Purkinje, granular layer and molecular layer neurons (a, right), midbrain neurons (a, left) and myenteric plexus neurons (b). NMDA receptor antibody binds to neural synapses primarily in hippocampus (c) and cerebellar granular layer (d). (see insert for color representation of the plate.)
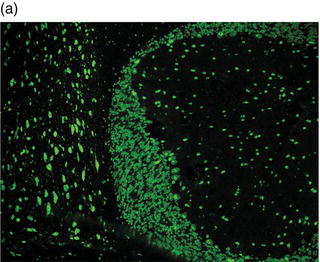
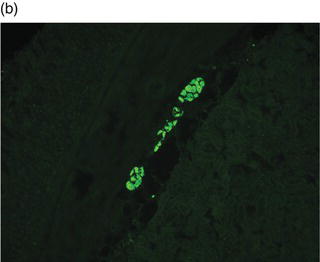
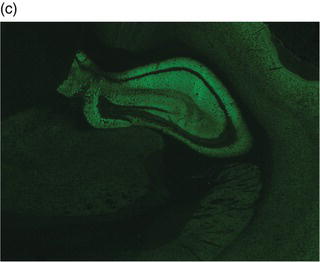
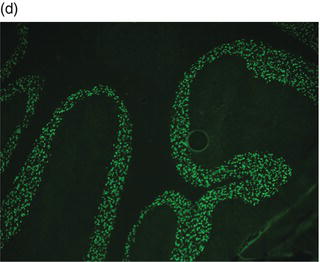
Given the diverse multifocal neurological accompaniments of patients with VGKC autoantibodies, patients who present with RPD frequently resemble neurodegenerative disorders, including frontotemporal dementia-like syndrome and CJD. Even when the clinical syndrome fulfills the diagnostic criteria for CJD, including cortical diffusion-weighted imaging hyperintensities, VGKC autoimmunity should be ruled out.
Recently, it was reported that target antigens in many patients with VGKC antibodies are proteins structurally and functionally associated with the channel. These antibodies target leucine-rich, glioma-inactivated 1 (Lgi1) and contactin-associated protein 2 (Caspr2) proteins. Clinical manifestations reported in association with Lgi1-IgG are LE (including presentation with RPD mimicking CJD), myoclonus, faciobrachial dystonic seizures, epilepsy, and hyponatremia. Caspr2-IgG has been described in patients with encephalitis, peripheral nerve hyperexcitability or both. Patients with encephalitis have been described to have subacute progressive memory difficulties, personality changes, and seizures.
Table 2.1 Neural autoantibodies targeting intracellular antigens reported to present with cognitive impairment, accompanying neurological findings and oncological associations
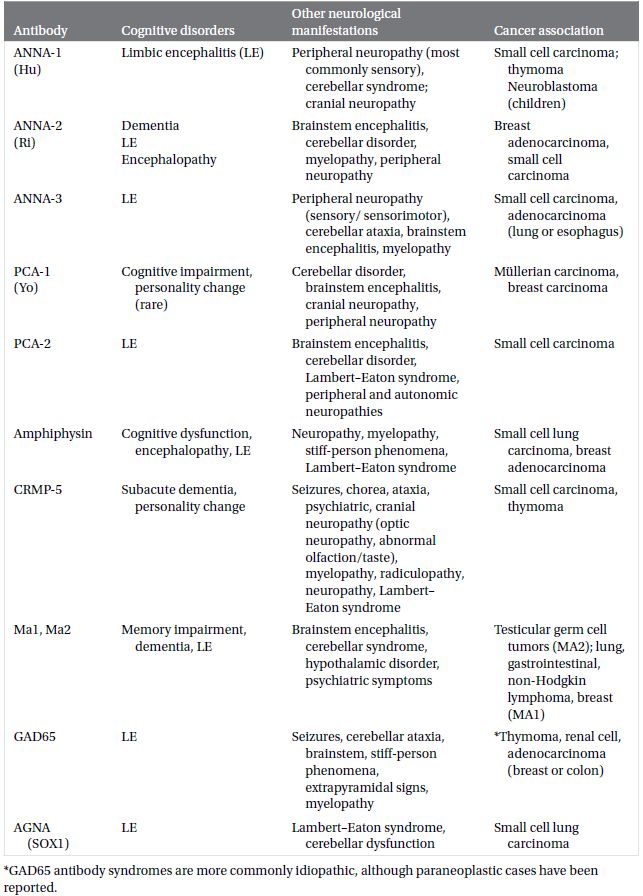
Table 2.2 Neural autoantibodies targeting cell membrane antigens reported to present with cognitive impairment, accompanying neurological findings and oncological associations
NMDA receptor antibody encephalitis
Autoantibodies to the N-methyl D-aspartate receptor (NMDAR) were first identified in 12 women with memory and psychiatric disturbances, decreased consciousness, and ovarian teratoma in 2007. NMDAR-IgG targets the NR1 (GluN1) subunit of the NMDAR, an ionotropic glutamate-gated cation channel that plays an important role in synaptic plasticity and excitotoxicity, mediating cognition, behavior, motor, respiratory, and autonomic control. This disorder is probably one of the most frequent among the neurological autoimmune encephalopathies.
Patients who develop rapidly progressive cognitive deterioration in NMDAR antibody encephalitis (NMDARAE) usually have a distinctive constellation and symptom evolution. Typically, the illness begins with prodromal symptoms, comprising fever, headache, upper respiratory and gastrointestinal symptoms, before patients develop psychiatric symptoms and memory impairment. One or more of seizures, abnormal movements (especially orofacial dyskinesias), autonomic instability, hypoventilation, and coma may follow.
While the distribution of patients with NMDARAE is highest in the young adult age groups, children and the elderly may also be affected. Female sex, 20s–40s age group, and African American race are risk factors for the detection of tumor, typically ovarian teratoma. Seventy-five percent of patients recover from this severe illness with immunotherapy and tumor removal (if present), although the recovery process is protracted and neurological dysfunction may persist, especially if treatment is delayed. Up to 25% of patients relapse, and risk for relapse increases in patients with no prior immunotherapy at onset, undetected tumor or rapid immunotherapy taper.
Diagnostic approach
In a patient with suspected autoimmune RPD, the diagnostic work-up is directed to finding objective evidence to support this diagnosis (clinically, radiologically, and on serum or CSF) and cancer detection. Parallel work-up to rule out other causes of rapidly progressive cognitive decline, especially those that are reversible, should also be performed. Brain imaging, EEG, and neuropsychometric testing can also provide baseline, objective parameters that can be followed over time as the patient receives treatment, to help evaluate treatment efficacy.
Autoantibody testing
Comprehensive neural autoantibody testing should be performed on serum and CSF. Selective isolated antibody testing is not advised, as the range of autoantibodies associated with acute cognitive decline is diverse and autoantibodies may co-exist. The neural antibody profile facilitates the identification of a paraneoplastic etiology (see Tables 2.1 and 2.2). An important caveat is that non-detection of a neural antibody does not exclude the diagnosis of autoimmune encephalitis when other clinical clues are evident.
Related posts:
 Diagnosis and Differential Diagnosis of Dementia
Diagnosis and Differential Diagnosis of Dementia
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


