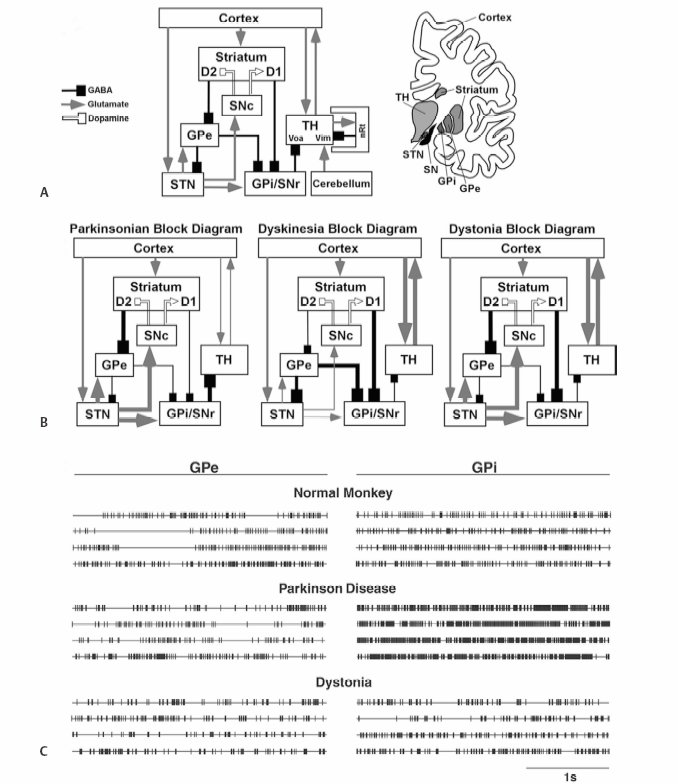
2 Rationale for Movement Disorder Surgery Cameron C. McIntyre, Christopher R. Butson, Benjamin L. Walter, and Jerrold L. Vitek Surgical interventions for movement disorders have been practiced for decades, beginning with early studies that used lesions to eliminate activity in localized brain regions. Later, deep brain stimulation (DBS) grew out of observations during such surgery. Surgeons using stimulating/ recording electrodes for target confirmation during ablative surgery found that stimulation of the brain had effects similar to lesioning, specifically that high frequency stimulation (> 100 Hz) of the motor thalamus produced tremor arrest.1 These early studies eventually proved that functional neurosurgery for movement disorders can alleviate symptoms and improve the quality of life for disabling diseases such as Parkinson disease (PD), essential tremor (ET), and dys-tonia in appropriately selected patients. Although these surgeries were initially based on empirical observations, the advancement of scientific understanding and medical technology has allowed for an evolution from empirically to rationally based surgical strategies. Current surgical approaches incorporate magnetic resonance imaging (MRI)-based stereotactically and neurophysiologically guided targeting where the selection of the target(s) is based on pathophysiological understanding of the disorder. Of the three major movement disorders for which surgical efficacy has been demonstrated (PD, ET, and dystonia), each has significant differences in the underlying pathophysiology. The cardinal symptoms of PD include resting tremor, rigidity, bradykinesia, and akinesia, as well as postural and gait disturbances.2,3 The pathological hallmark of PD is the degeneration of dopaminergic cells within the substantia nigra with deposition of cytoplasmic eosinophilic inclusions (Lewy bodies) and subsequent dopamine depletion of the striatum. Clinical manifestations of PD occur when nigrostriatal dopamine loss reaches ~60%.4 Dopamine denervation of the striatum triggers a cascade of events within the basal ganglia that impairs its ability to appropriately interact with the rest of the nervous system. Medical treatment of PD with levodopa is not the panacea it was initially thought to be due to the late complications of levodopa-induced dyskinesias and increased motor fluctuations. Even with molecular and genetically based therapies on the horizon aimed at slowing the progression of PD, there is and will be for the foreseeable future a large population of patients significantly burdened by this neurodegenerative disease. Approximately 4% of the population > 40 years of age exhibit symptoms of ET.5,6 Most ET patients have mild symptoms that can be controlled with medication. However, a significant number of patients are refractory to medication, have significant disability, and are unable to perform simple activities of daily living or maintain employment. Although the neurophysiological origin of tremor remains debated within the scientific community, there remains a sustained need to address the limitations of current pharmacological approaches to the treatment of ET. Dystonia is characterized by sustained muscle contractions, leading to twisting, repetitive movements, and abnormal postures. It can be focal, involving a single body region, segmental, involving two or more adjacent regions, or generalized, involving both legs plus at least one other region. Dystonia can be a symptom of destructive disease processes and in such cases is termed secondary dystonia. Dystonia with no obvious pathology is called primary dystonia. Some of these patients have now been found to have an inherited gene mutation, and there are now over 14 gene loci identified in association with dystonia. DYT1 dystonia, the most common of these, typically starts focally with one limb in early childhood and slowly generalizes to involve other body regions. Other genetic loci responsible for primary generalized dystonia include DYT2, 4, 6, 7, and 13.7 Reports of neuropathological examination of patients with suspected primary generalized dystonia are limited, and standard MRI scans have been reported as normal. Until recently no neuropathological changes have been reported in the brains of DYT1 patients.8 However, a recent study found perinuclear inclusion bodies that stain positive for ubiquitin, torsinA, and lamin A/C in the pedunculopontine nucleus, cuneiform nucleus, and griseum centrale mesencephal.9 Clinicopathological studies in patients with secondary dystonia indicate that the most common site in which a lesion can be identified is in the striatum, or pallidum.10 Thalamic lesions, although less common, have also been observed in patients with dystonia.11 Dystonia is a particularly disabling condition, and although many patients with focal dystonias involving small muscle groups may respond to botulinum toxin, others with more diffuse involvement or involvement of larger muscle groups have limited success with traditional medical therapies. PD, ET, and dystonia each represent especially debilitating diseases that affect significant numbers of patients who do not adequately respond to pharmacological interventions. In turn, functional neurosurgery has been employed in each disease state with substantial degrees of success. The combination of basic science understanding, advancing medical technology, and clinical acceptance has allowed the development of successful surgical practices and clinical centers. Currently there are two forms of surgical intervention for movement disorders that are used most commonly (ablation and DBS), and three general anatomical targets [thalamus, subthalamic nucleus (STN), and globus pallidus (GP)] to which these approaches are applied. Thalamic DBS for intractable tremor has virtually replaced ablative lesions of the thalamus.12 Similarly, DBS of the STN or internal segment of the globus pallidus (GPi) has largely replaced pallidotomy in the treatment of the cardinal motor features of PD (resting tremor, rigidity, bradykinesia).13 In addition, multiple pilot studies have begun to examine the utility of DBS for dystonia,14,15 epilepsy,16 and obsessive—compulsive disorder (OCD).17 As data from larger controlled trials become available, neurologists and neurosurgeons will be able to tailor the therapy for their patient based on the patient’s disease characteristics (i.e., appropriate choice of target site and mode of treatment, ablation, or stimulation). Furthermore, we will be able to better predict the degree of clinical benefit and risk of adverse side effects based on characteristics of the patient’s disease, age, and other clinical variables. The basal ganglia (BG) are located in the basal telencephalon and consist of four interconnected nuclei: the striatum, GP, substantia nigra, and STN (Fig. 2.1). The thalamus is a large, oval-shaped structure that constitutes the dorsal portion of the diencephalon and acts as the gateway to the cortex. All sensory pathways (except olfaction) relay in the thalamus en route to the cerebral cortex as well as many of the anatomical circuits of the cerebellum, BG, and limbic structures. The striatum is the main input structure of the BG; the GPi and the substantia nigra pars reticulata (SNr) are the output nuclei.18 Glutamatergic projections from virtually all cortical areas, as well as dopaminergic inputs from the substantia nigra pars compacta (SNc), converge onto spiny projection neurons in the striatum. Striatal output typically consists of low-frequency activity (~10 Hz), which is transmitted to the output nuclei (GPi and SNr) via either a direct or an indirect pathway.18–20 In the direct pathway, one subpopulation of spiny neurons projects directly to the output nuclei (GPi and SNr) of the BG. A separate subpopulation of striatal neurons follows an indirect pathway, first projecting to the external segment of the globus pallidus (GPe), which projects to the STN, which in turn sends projections to the GPi and SNr. The subpopulations of spiny neurons that give rise to the direct and indirect pathways are characterized by their selective expression of neuropeptide and dopamine receptor subtypes.21 Although all striatal spiny neurons use gamma-amino butyric acid (GABA) as their main neurotransmitter, the subpopulation that gives rise to the direct pathway preferentially express the D1 subtype of the dopamine receptor (primarily excitatory), and the subpopulation that gives rise to the indirect pathway preferentially express the D2 subtype of the dopamine receptor (primarily inhibitory).20 In turn, dopaminergic inputs from the SNc can explicitly modulate the transmission of neural activity through the direct and indirect pathways (Fig. 2.1). The GPe, GPi, and SNr are each composed primarily of projection neurons that typically exhibit a consistent high-frequency (~60 Hz) firing pattern and use GABA as their main neurotransmitter. The inhibitory GPe output is transmitted to the GPi and STN. The STN is composed primarily of projection neurons that typically exhibit a consistent midfrequency (~40 Hz) firing pattern and use glutamate as their main neurotransmitter. Excitatory STN output makes a feedback loop with GPe and also projects to GPi and SNr. In addition, the STN receives direct excitatory cortical inputs that can modulate its firing rate and pattern. The direct and indirect pathways both project to the GPi and SNr, which sends inhibitory projections to the thalamus (Fig. 2.1). By virtue of the neurotransmitters and baseline activity of neurons in the BG network, activation of the direct or indirect pathways can produce functionally opposite effects on thalamic output.20 Corticostriatal neurons, thalamocortical neurons, and neurons of the STN are excitatory, utilizing glutamate as a neurotransmitter. All other neurons in the BG network are inhibitory, using GABA as their main neurotransmitter. Under resting conditions, the activity of the output neurons of the striatum is low compared with that of tonically active neurons in the GPe and STN. Activation of the corticostriatal pathway leads to increased firing of striatal neurons. Increased activity of striatal neurons in the direct pathway (striatum → GPi/SNr) leads to inhibition of the output nuclei (GPi and SNr). A reduction in tonic activity of the neurons in GPi/SNr leads to a reduction in the inhibition of neurons in the thalamus and increased thalamocortical output. In contrast, activation of the indirect pathway (striatum → GPe → STN → GPi/SNr), leads to the opposite functional effect on the thalamus. Increased activity of the striatal output neurons inhibits the tonically active neurons in the GPe. Inhibition of the neurons in GPe disinhibits neurons in the STN. Increased activity of the excitatory neurons of the STN leads to increased firing of neurons in GPi and SNr. An increase in the tonic activity of the neurons in GPi and SNr leads to an increase in the inhibition of neurons in the thalamus and a reduction in thalamocortical output (Fig. 2.1). The thalamus is composed of nuclei most commonly classified into four groups (anterior, medial, ventrolateral, and posterior) based on the anatomical divisions created by the internal medullary lamina. The ventrolateral thalamus constitutes nearly all the nuclei intimately involved in sensorimotor function. The motor thalamus can be further subdivided into distinct relays for BG and cerebellar afferents. These channels in turn gain access to different functional regions of the cerebral cortex.22 Anatomical tracing studies in nonhuman primates show that pallidal and cerebellar receiving areas in the thalamus are largely segregated. GPi projects to the anterior part of the ventrolateral thalamus (human—Voa/Vop; monkey—VLo), which predominantly projects to the supplementary motor cortex. The cerebellum predominantly projects to the posterior part of the ventrolateral thalamus (human—Vim/Voi; monkey—VPLo), which then projects to the motor cortex (area 4) and arcuate premotor area.23,24 Fig. 2.1 Basal—ganglia—thalamo—cortical network. (A) Simplified circuit diagram of the basal ganglia. Relative thickness of the lines indicates degree of activation, arrows represent excitatory synapses, squares represent inhibitory synapses, and line color indicates neurotransmitter [white, dopamine; gray, glutamate; black, gamma-amino butyric acid (GABA)]. Cortical information that reaches the striatum is conveyed to the basal ganglia output structures [internal segment of the globus pallidus (GPi) and the substantia nigra pars re-ticulata (SNr)] via two pathways, a direct inhibitory projection from the striatum to the GPi/SNr, and an indirect network that involves an inhibitory projection from the striatum to the external segment of the globus pallidus (GPe), an inhibitory projection from the GPe to the subthalamic nucleus (STN), an inhibitory projection from the GPe to the GPi/SNr, and an excitatory projection from the STN to the GPi/SNr. The information is then transmitted back to the cerebral cortex via a relay in the thalamus. (B) In Parkinson disease, the dopaminergic cell loss of the substantia nigra pars compacta (SNc) causes a cascade of alterations affecting all the other components of the circuit. The final result is the increased activity of the GABAergic output nuclei GPi/SNr, which causes increased inhibition of the motor thalamus. By contrast, hyperkinetic disorders such as dystonia result in increased inhibition of GPi/SNr, resulting in decreased inhibition of thalamus and overactivity of thalamic projections to cortex. (C) Examples of rasters of spontaneous neuronal activity in a normal monkey (top), a patient with Parkinson disease (PD) (middle), and a patient with dystonia (bottom). These illustrate the firing rates in GPi are reduced in dystonia and increased in PD.93,94 In both PD and dystonia there are abnormal, irregular grouped discharges in GPi, whereas in normal primates GPi has a more tonic regular firing pattern.93 (Sources: Alexander GE, Crutcher MD, DeLong MR. “Basal ganglia—thalamocortical circuits: parallel substrates for motor, oculomotor, “prefrontal” and “limbic” functions.” In: Uylings HBM, Van Eden CG, De Bruin JPC, Corner MA, Feenstra MGP, eds. Progress in Brain Research. New York: Elsevier Science Publishers; 1990 and Smith Y, Bevan MD, Shink E, Bolam JP. Microcircuitry of the direct and indirect pathways of the basal ganglia. Neuroscience 1998;86:353–387.) The majority of neurons within the various thalamic nuclei are thalamocortical projection neurons that use glutamate as their primary neurotransmitter. These neurons typically exhibit a relatively low frequency tonic firing pattern (~20 Hz). However, thalamic neurons transition from a tonic firing pattern to a low-frequency burst firing pattern (~4 to 8 Hz interburst interval) dependent on the behavioral state, which determines the synaptic input conditions.25,26 Another major component of the thalamic network consists of a thin sheet of inhibitory neurons, known as the reticular nucleus (nRt). The nRt forms the anterior lateral and a portion of the dorsal surfaces of the thalamus. Reticular neurons provide widespread inhibitory input to cells in the entire thalamus. In addition, thalamocortical projection neurons provide direct inputs to reticular thalamic neurons, generating tightly linked feedback loops. In turn, the interaction of the thalamocortical and reticular neurons provides an additional degree of information processing within both motor and nonmotor circuits of the thalamocortical system27 (Fig. 2.1). The rate-based block diagram of the BG—thalamocortical network is a highly Simplified representation. The feedback interaction between the STN and GPe,28,29 direct cortical inputs to the STN,30 and the interaction of additional nuclei such as the pedunculopontine nucleus31 and reticular thalamus20 complicate our understanding of this network. In addition, this model does not take into account the pattern and/or synchrony of neural activity in each of the interacting nuclei. Although the rate-based model does not capture all of the important details pertinent to this understanding of the pathophysiological basis underlying the development of hypokinetic and hyperkinetic movement disorders, it has provided a great deal of scientific and clinical insight that has led to testable hypotheses and the development of alternate models that take these new observations into account. Our understanding of the pathophysiological basis of hypo- and hyperkinetic movement disorders has increased significantly over the last decade. The physiology of the normal brain relative to hyper- and hypokinetic disease states is summarized in Fig. 2.1, which highlights the neuronal activity changes that take place in the BG and thalamus in these disease states. Sensorimotor, associative, oculomotor, and limbic circuits originate in specific cortical areas, pass through distinct portions of the BG and thalamus, and project back to their cortical area of origin. These circuits are anatomically and functionally segregated throughout the BG—thalamo—cortical network, and disruption of the motor circuit is of primary interest in analysis of movement disorders. Both neuronal firing rates and patterns of activity within the BG network are now considered important in the genesis of movement disorders. In addition, the contribution of altered receptive fields and changes in synchrony are just now being appreciated. These scientific advances are providing a rationale for the selection of anatomical targets for ablative or DBS movement disorder surgery. The pathophysiological basis underlying the development of PD has been developed from electrophysiological studies in animal models of PD as well as from humans with idiopathic PD undergoing functional neurosurgical procedures. The loss of dopamine from the SNc in PD is proposed to lead to differential changes in neuronal activity of striatal cells in the direct and indirect pathway. In the direct pathway, loss of dopamine at striatal excitatory D1 receptors leads to a decrease of inhibitory activity from the striatum to GPi. In the indirect pathway, there is loss of dopamine at inhibitory D2 receptors leading to increased activity of inhibitory striatal neurons projecting to GPe causing a reduction of GPe activity. The decrease in inhibitory output from GPe to STN leads to excessive excitation from the STN to the GPi. Thus there is an increase in inhibitory activity from the GPi to the thalamus and brain stem, via both the direct and the indirect pathways (Fig. 2.1B), which has been implicitly implicated as the change in neuronal circuits that underlies the development of the hypokinetic features associated with PD.32 Changes in mean firing rates of neurons in GPe, GPi, STN, and VLo in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) monkey model of PD, and in GPe and GPi in patients with idiopathic PD, show a decrease in mean discharge rates in GPe and VLo and an increase in mean discharge rates in STN and GPi.32–35 These changes are consistent with predictions from the rate-based block diagram of PD. Similarly, a loss or lowering of inhibitory input from GPi to the thalamus has been proposed as the basis for the development of the hyperkinetic movements associated with drug-induced dyskinesias and other hyperkinetic disorders (e.g., dystonia and hemiballismus). Changes in neuronal firing rates alone, however, are not sufficient to explain the pathophysiology of PD or hyperkinetic movement disorders. Lesions within the motor thalamus do not exacerbate or induce parkinsonian motor signs, but instead, are reported to improve or abolish parkinsonian tremor, rigidity, and drug-induced dyskinesias.36,37 Similarly, lesions in the GPi do not induce hyperkinetic movement disorders, but instead have been reported to abolish drug-induced dyskinesias as well as the involuntary movements associated with dystonia and hemiballismus. These results suggest that a decrease in activity of thalamicneurons cannot by itself account for the development of parkinsonian motor signs, nor can a reduction in the rate of GPi neurons account for the development of drug-induced dyskinesias, dystonia, or hemiballismus.38,39 These contradictions of the rate model have led to the incorporation of additional hypotheses to explain the pathophysiology of hypokinetic and hyperkinetic movement disorders. In addition to the changes in neuronal firing rate, movement disorders are also associated with altered patterns of neuronal activity, changes in somatosensory responsiveness, and the degree of synchrony of neurons within the BG and thalamus (Fig. 2.1). The physiological basis for the development of PD has been a much debated issue. However, it appears likely that PD motor symptoms arise from a combination of changes in both rate and pattern. Observations of increased bursting and rhythmic oscillatory patterns within the BG and thalamus are common in both parkinsonian monkeys and humans with PD.19,40 Bursting activity of thalamic neurons in the cerebellar receiving area of the thalamus is strongly correlated with tremor in PD, as well as ET.41 In addition, oscillatory activity, changes in somatosensory responsiveness, and degree of synchronization of pallidal and STN neurons are considered prominent features of PD pathophysiology.29 Pathophysiologically, dystonia can be viewed as either a hypo- or hyperkinetic movement disorder. Dystonia may be a presenting symptom in parkinsonian patients who are not taking antiparkinsonian medications (“off”-dystonia)or alternatively may appear as a consequence of L-dopa treatment (peak dose dystonia). Electrophysiological recordings of pallidal neurons in patients with idiopathic dystonia demonstrate a decrease in the mean discharge rates of GPi neurons, consistent with predictions based on the “rate” model for hyperkinetic movement disorders.42–44 However, in addition to decreased mean discharge rates, neuronal activity in the BG of dystonic patients have also demonstrated altered patterns of activity and widened receptive fields.44 Uncontrolled changes in synchronization of neuronal populations in GPi may also occur and have been suggested to play an important role in the development of dystonia.45 However, the exact relationship between changes (rate and pattern) in neural activity and the development of dystonia remains unclear. The clear clinical improvements in movement disorder symptoms after thalamotomy, subthalamotomy, pallidotomy, or DBS are difficult to reconcile based solely on regularization of firing rate because each intervention should result in a different effect on rate. Therefore, given the pathophysiological characteristics of neuronal firing patterns in movement disorders, it has been suggested that the therapeutic effects of the treatment are dictated primarily by effects on the firing pattern. Based on this hypothesis, both ablation and DBS are effective in the treatment of movement disorders because they replace pathological neuronal activity with either no activity (ablation) or a highly regular firing pattern (DBS).46–48
Anatomy and Physiology of the Basal Ganglia and Thalamus
Pathophysiological Basis of Movement Disorders
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


