Fig. 1
Preventive effects of intracerebroventricular injection of r-OPN on Evans blue dye extravasation (arrow, meaning BBB disruption) at 24 h after SAH by endovascular puncture. Sham, sham-operated rats treated with phosphate-buffered saline (PBS) vehicle; SAH or SAH-OPN, SAH rats treated with PBS or 0.1 μg of r-OPN
Nasal administration of r-OPN also decreased neuronal cell death and brain edema, and improved the neurological status in SAH rats by endovascular puncture, possibly through focal adhesion kinase-phosphatidylinositol 3-kinase-Akt-induced inhibition of capase-3 cleavage [23]. This study did not examine anti-apoptotic effects of r-OPN on capillary endothelial cells, which can potentially explain r-OPN’s inhibition of brain edema formation after SAH.
On the other hand, r-OPN suppressed post-SAH endogenous OPN induction in brain, although r-OPN had no effects on endogenous OPN levels in the sham-operated rats [13]. These findings may reflect the decreased brain damage by r-OPN, because OPN is induced in response to tissue injuries or inflammation and may play a role in the maintenance of tissue homeostasis and the induction of tissue repair in a variety of situations [3].
TNC
Expression of TNC in SAH Brain
In a clinical setting, TNC levels were increased in both serum and cerebrospinal fluid (CSF) after aneurysmal SAH [16–20]. CSF TNC levels peaked immediately after SAH and decreased with time, whereas serum TNC levels increased transiently and peaked on Days 4–6 [19]. Serum TNC levels were greater in patients with subsequent angiographic vasospasm; the peak occurred 2.4 days before an increase in the mean transcranial Doppler velocity to ≥120 cm/s and 3.6 days before the onset of delayed cerebral ischemia [18]. In contrast, CSF TNC levels were below the diagnostic threshold level in control patients and markedly increased after SAH [20]. Higher CSF TNC levels were observed in patients with worse admission clinical grade, more severe SAH on admission computed tomography, acute obstructive hydrocephalus, subsequent angiographic vasospasm, delayed cerebral ischemia, chronic shunt-dependent hydrocephalus, and a worse outcome [16, 17, 20]. To predict the onset of delayed cerebral ischemia, 16.2 ng/mL was considered as an appropriate cut-off value for CSF TNC on Days 1–6, giving a sensitivity of 81.0 % and a specificity of 79.5 % [16]. Clinical findings suggest that more severe SAH or initial brain injury may induce more TNC, which may cause angiographic vasospasm and delayed cerebral ischemia separately or simultaneously; that is, delayed cerebral ischemia may occur by severe angiographic vasospasm with more TNC induction, and/or by vasospasm-unrelated causes with TNC induction, which is thought to be early brain injury [17].
TNC is induced in both cerebral arterial wall [12] and brain parenchyma after experimental SAH by endovascular puncture in rats [11]. TNC induction in the cerebral artery caused vasospasm [4, 12]. On the other hand, TNC was increased in neuropil mainly in the brain surface of the cerebral cortex irrespective of cerebral vasospasm development at both 24 and 72 h after SAH [11]. It was considered that astrocytes, neurons, and capillary endothelial cells produced TNC [11].
Role of Endogenous TNC Induction in BBB Disruption after SAH
Because there are neither inhibitors nor neutralizing antibodies specific for TNC, a previous study used imatinib mesylate (a selective inhibitor of the tyrosine kinases of platelet-derived growth factor [PDGF] receptors [PDGFRs]) to block endogenous TNC induction and to examine the effects on brain injury after SAH by endovascular puncture in rats [11]. PDGF is well known to be a potent inducer of TNC [12]. Actually, imatinib mesylate, a PDGFR inhibitor, prevented brain PDGFR activation, TNC upregulation, MAPK activation, neuronal apoptosis, and neurological impairments after SAH in rats (Fig. 2), although the effects on endothelial cell apoptosis or brain edema formation were not examined [11]. Another study reported that imatinib mesylate prevented post-SAH BBB disruption and brain edema formation by inhibiting JNK-mediated MMP-9 activation in rats [26]. In a clinical setting, TNC was observed around hyperplastic blood vessels of astrocytomas, regardless of their grade, as well as around newly formed vascular channels of inflammatory, ischemic, and traumatic diseases of the brain, suggesting the role of TNC in angiogenesis [25]. Taken together, TNC induction in SAH brain may cause BBB disruption and brain edema formation, although the possibility that imatinib mesylate exerts neuroprotective effects by TNC-unrelated mechanisms cannot be excluded. Our preliminary study also showed that TNC-knockout mice had a significant decrease in post-SAH BBB disruption and brain edema formation associated with MMP-9 inhibition (unpublished data).
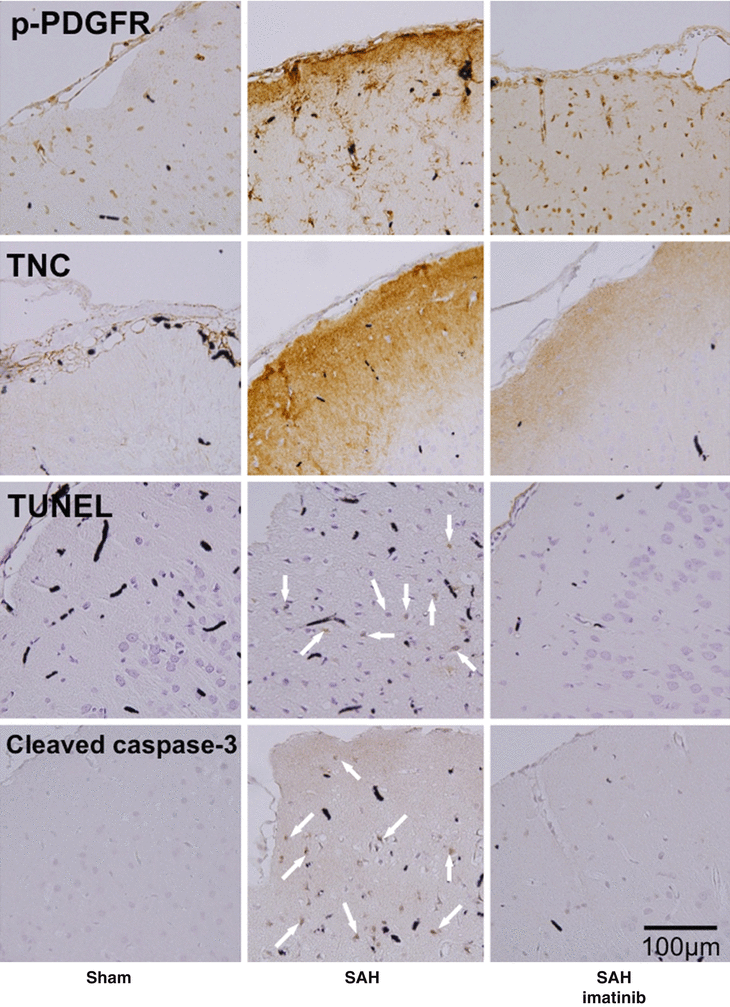
Fig. 2
Inhibitory effects of intraperitoneal injection of imatinib mesylate on expression of phosphorylated PDGFR (p-PDGFR), TNC, and cleaved caspase-3, or terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL) staining in brain at 24 h after SAH by endovascular puncture. Arrow, apoptotic neuron; SAH-imatinib, SAH rats treated with imatinib mesylate (50 mg/kg body weight). Black stains depict India ink, because specimens were harvested after India ink angiography
Effects of Exogenous TNC on BBB Disruption after SAH
A cisternal injection of intact (full-length) TNC induced prolonged cerebral arterial constriction but did not cause neurological impairments in healthy rats [4], whereas a cisternal injection of recombinant TNC (r-TNC; murine myeloma cell line, NS0-derived, Gly23–Pro625, with a C-terminal 6-His tag) caused vasospasm, neuronal apoptosis, and neurological impairments in imatinib mesylate-treated filament puncture SAH rats [11, 12]. Exogenous TNC activated MAPKs in the cerebral artery and caused cerebral arterial contraction in both healthy rats and imatinib mesylate-treated SAH rats [4, 11, 12]. However, effects of exogenous TNC on brain may be different between healthy and SAH rats, because SAH may induce MMPs, which can cleave TNC. Cleaved TNC may activate different signaling and exert diverse cell responses depending on domains of TNC [9]. Our recent preliminary study showed that a cisternal injection of intact TNC aggravated BBB disruption and brain edema in TNC-knockout filament puncture SAH mice (unpublished data).
An interesting feature of TNC is the existence of positive feedback mechanisms to augment TNC’s actions [4, 11, 12]. A r-TNC injection induced TNC itself in the cerebral artery and brain after SAH, which may internally augment vasospasm and neuronal apoptosis [11, 12]. After SAH, PDGF may induce endogenous TNC, and r-TNC also induced both PDGFR-β upregulation and PDGFR activation [12]. These findings suggest that PDGF-induced TNC may positively feed back on PDGFR activation via PDGFR upregulation and crosstalk signaling between receptors as well as upregulation of TNC, leading to more MAPK activation and therefore cerebral vasospasm or neurological impairments in SAH rats [12]. Another study using healthy rats showed that a cisternal intact TNC injection induced prolonged cerebral arterial constriction via toll-like receptor 4 (TLR4) and activation of MAPKs, and suggested that TLR4 activation may have positive feedback on TLR4 activation via MAPK-mediated upregulation of TNC and TLR4 in cerebral arteries, causing more activation of MAPKs and more prolongation of vasoconstriction, even though post-injection TNC concentration in CSF decreases over time [4].
Discussion
The biological functions of MCPs are highly variable and often seemingly contradictory, depending on the biological scenario surrounding their induction [13]. However, OPN is consistently protective [13, 15], whereas TNC is deleterious for brain or BBB after SAH [11, 17, 20]. It is notable that the function of OPN and TNC, representatives of MCPs, is conflicting in the setting of SAH. As to cerebral vasospasm after SAH, r-OPN activated the protective pathways including MKP-1, an endogenous MAPK inhibitor, via binding to RGD-dependent integrins [14]. In contrast, exogenous TNC activated MAPKs via binding to TLR4 and caused cerebral vasospasm or prolonged cerebral artery contraction [4, 12]. Although the mechanisms of how OPN antagonizes TNC’s effect remain unclear in cerebral vasospasm after SAH, another possibility is that OPN may inhibit TNC’s binding to its receptor competitively, because they share some receptors [21]. TNC signaling may positively feed back on upregulation of TNC itself in an acute phase, leading to more activation of the signaling transduction and the development of cerebral vasospasm [12], while OPN has no such action and seems to be induced in a delayed fashion in response to tissue injuries or inflammation [3, 13].
Related posts:
 of Behavioral Deficits in Rodents Following Brain Injury Across Species, Gender, and Experimental Model
of Behavioral Deficits in Rodents Following Brain Injury Across Species, Gender, and Experimental Model
 Infarction After Aneurysmal Subarachnoid Hemorrhage
Infarction After Aneurysmal Subarachnoid Hemorrhage
 Volume Determination in Subarachnoid Hemorrhage Using Rats
Volume Determination in Subarachnoid Hemorrhage Using Rats
 Pretreatment Fails to Provide Neuroprotection Following a Surgically Induced Brain Injury Rat Model
Pretreatment Fails to Provide Neuroprotection Following a Surgically Induced Brain Injury Rat Model
 IGF-1 Reduced Rat Pup Germinal Matrix Hemorrhage
IGF-1 Reduced Rat Pup Germinal Matrix Hemorrhage
 Injection of Noncellular Cerebrospinal Fluid from Subarachnoid Hemorrhage Patient into Rat Ventricles Leads to Ventricular Enlargement and Periventricular Injury
Injection of Noncellular Cerebrospinal Fluid from Subarachnoid Hemorrhage Patient into Rat Ventricles Leads to Ventricular Enlargement and Periventricular Injury
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


