Fig. 1
A scheme of integration of biotechnologies and applications for development of personalized medicine
The role of neurogenomics in the development of personalized neurology is shown schematically in Fig. 2. Only important relationships are shown. Genomic neurology is not an appropriate synonym for personalized neurology as some cell therapies do not require a knowledge of the human genome and several biomarkers to guide personalized treatment may be biochemical rather than genomic. Personalized medicine, however, existed long before the advent of genomic age. Ancient medical systems such as Ayurveda and Chinese medicine were personalized taking into consideration variations among patients with the same disease.
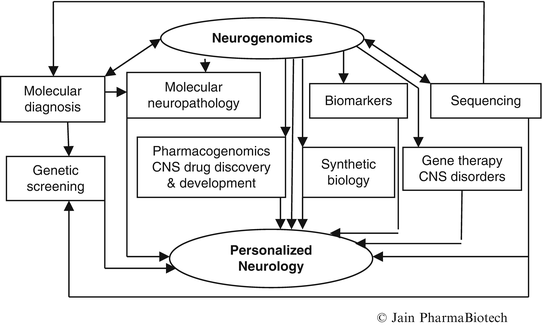
Fig. 2
Role of neurogenomics in the development of personalized neurology
The combination of genomic, proteomic, glycomic, and metabolomic approaches may yield novel insights into molecular mechanisms of disease pathophysiology, which could then be integrated and translated into clinical neurology (Gotovac et al. 2014). This article will focus on the role of neurogenomics in the development of personalized neurology.
2 Personalized Approach to Individual Neurological Disorders
This section will describe personalized management of important neurological disorders. Since the focus of this chapter is on the role of neurogenomics, non-genomic aspects will not be discussed. However, because personalized neurology involves integration of several technologies, many of which also interact with genomics, it is impossible to exclude other technologies in a personalized approach to a particular neurological disorder. Examples of the application of neurogenomic technologies leading to the development of personalized neurology include Alzheimer disease, Parkinson disease, multiple sclerosis, traumatic brain injury, migraine, epilepsy, and pain and chronic fatigue syndrome.
2.1 Alzheimer Disease
The interaction of different transcription factors with the regulatory region of the ApoE gene plays an important role in the neuroinflammatory process seen in Alzheimer disease (AD) and is a target for developing new therapeutics for the disease. Genotype-specific responses of AD patients to a particular drug or combination of drugs have been demonstrated, although several studies examining the role of ApoE produced conflicting results. The pharmacogenomics of Alzheimer disease may, in the future, contribute to optimizing drug development and therapeutics, increasing efficacy and safety, and reducing side effects.
Associations between the GAB2 gene and late-onset AD (LOAD) risk have been characterized in ApoE 34 carriers by a genome-wide survey of >300,000 SNPs (Reiman et al. 2007). The discovery of this LOAD susceptibility gene, if replicated, provides new opportunities to investigate LOAD pathogenesis, predisposition, treatment, and prevention. Genome-wide studies using even higher density platforms and compound genetic analyses in sufficiently large samples of well-characterized cases and controls promise to play increasingly important roles in the scientific understanding, evaluation, personalized treatment, and prevention of AD.
Apart from ApoE, ~20 genes are associated with AD. One of these, SORL1, was discovered during an international study that analyzed DNAs from >6,000 persons from an isolated population in the Dominican Republic. The study found that this gene can raise the risk of developing AD three times in this population (Rogaeva et al. 2007). Another identified gene, CALHM1, encodes the essential component of a cerebral Ca2+ channel that controls Aβ levels and susceptibility to late-onset AD, suggesting a potentially new way to treat or even prevent the disease (Dreses-Werringloer et al. 2008).
Genome-wide scans were used to screen the brain’s connectivity pattern and the SPON1 variant at rs2618516 on chromosome 11 (Jahanshad et al. 2013). Older persons who carried the connectivity variant rs2618516 had significantly milder clinical dementia scores.
2.2 Parkinson Disease
Results of the largest case-control genome-wide association study so far indicate a substantial contribution of genetics to susceptibility for both early onset and late-onset Parkinson disease (PD), although most of the genetic components of this disease remain to be discovered (Do et al. 2011).
Levodopa is used to titrate dopamine up to an optimal level for movement and some aspects of cognition. Response of individual patients and adverse reactions vary considerably. An understanding of the basis of these differential effects may enable modification of the drug dose, or combination of levodopa with other drugs, to produce the best outcome for individual patients and to avoid such reactions. There is a trend now toward incorporating genetics into clinical studies of therapy for PD to investigate how a person’s genetic makeup influences the effect of drugs that work by neurochemical intervention. Approximately 50 % of PD patients treated with l-dopa develop l-dopa-induced dyskinesias in the long term, and the use of pharmacogenetics should be explored in an effort to reduce this complication.
2.3 Multiple Sclerosis
Genetic factors are considered to be responsible for the increased frequency of the disease seen in the relatives of individuals affected with multiple sclerosis (MS). Genome-wide association studies have identified several risk loci, and the variation within the major histocompatibility complex (MHC) exerts the greatest individual effect on risk. MHC in chromosome 6p21.3 represents by far the strongest MS susceptibility locus genome-wide and was unambiguously identified in all studied MS populations (Oksenberg 2013). Immunologically relevant genes are overrepresented among those mapping close to the identified loci and implicate T-helper cell differentiation in the pathogenesis of MS (International Multiple Sclerosis Genetics Consortium et al. 2011).
Pharmacogenetic research models based on a high-throughput single nucleotide polymorphism (SNP) technology have been used to establish the correlation between drug responsiveness and genetic polymorphisms of MS patients, which may promote the development of personalized medicine for this disease. Spectratyping has shown that T cells bearing particular types of receptors are activated in MS patients. T-cell receptor-based immunotherapy can be applicable to MS patients if the T-cell receptor activation pattern of each patient is determined at different stages of the disease. The BEST-PGx (Betaferon/Betaseron in Early relapsing-remitting MS Surveillance Trial-Pharmacogenomics) has investigated the value of RNA expression profiling and pharmacogenetics in predicting the treatment response to interferon beta in patients with early relapsing MS (Kappos et al. 2005).
Genome-wide expression studies in brain tissue and blood samples of MS patients are expected to reveal biomarkers that would help to determine disease course, outcome, or treatment response in early stages of the disease (Habek et al. 2010). An increasing number of genetic polymorphisms have been correlated with MS, but so far their relevance to diagnosis of MS is rather low. A large number of genes (including GSTM, IL1B, PD-1, CCR5, OPN, IL4, HLA-DRB1*1501, CD24, ESR1, CD59, CNTF, CRYAB, IFNγ, MEFV, APOE, TGFB1) have been associated with certain MS phenotypes, but these correlations are often controversial. Research on pharmacogenomics of MS is increasing but has not produced a useful biomarker for clinical practice so far (Comabella and Vandenbroeck 2011).
2.4 Traumatic Brain Injury
There is a considerable variation in the response of patients to traumatic brain injury (TBI). The expression of some genes such as APOE has been implicated in outcome following TBI, but extensive review of literature has revealed contradictory results that are attributable to the heterogeneity of studies. Further research is needed to assess the relationship between genetic traits and clinical outcome of TBI (Davidson et al. 2014). Biomarkers are useful as diagnostic, prognostic, and monitoring adjuncts. Changes in the expression profile of biomarkers such as microRNAs in peripheral blood mononuclear cells may reflect molecular alterations following brain injury that contribute to the sequelae (Pasinetti et al. 2010).
Mild hypothermia, used for neuroprotection following TBI, has a significant effect on the gene expression profiles of the hippocampus (Feng et al. 2010). Differential expression of these genes may be involved in the mechanisms of neuroprotection.
2.5 Stroke
Stroke accounts for four and a half million deaths each year with an estimated nine million stroke survivors annually. The overall incidence rate of stroke is 2–2.5 per 1,000 adults with a prevalence of ~5 per 1,000 and an estimated 5-year risk of stroke recurrence of 15–40 %. Conventional risk factors for stroke include increasing age, hypertension, diabetes mellitus, smoking, increased body mass index, ischemic heart disease, heart failure, atrial fibrillation, and lack of physical activity. Age is the strongest risk factor for both ischemic and hemorrhagic strokes with its incidence doubling for each successive decade after the age of 55 years. However, there is a substantial portion of patients with significant cerebrovascular disease who do not have any of these stroke risk factors, and it may be helpful to identify complex genetic determinants such as multiple genes that play a role. There is no cure for stroke, but principle drugs used currently are antithrombotics, and their efficacy and safety can be improved by using pharmacogenetics and pharmacogenomics (Billeci et al. 2009). Personalization of stroke management should start at the stage of clinical trials of various therapies. Stroke treatments may be neuroprotective in the acute stage and neuroregenerative or neurorestorative in the subacute and chronic stages. Various biomarkers and brain imaging can be used to guide clinical trials. A number of factors are taken into consideration for personalizing treatment of stroke. An example of relevance to genomics will be mentioned here.
Intracerebral hemorrhage (ICH) usually has poor prognosis, which is not altered by surgical evacuation of the hematoma in most cases. Vasculopathic changes associated with the APOE ε2 allele might have a role in the severity and clinical course of lobar ICH. Screening of patients who have ICH to identify the ε2 variant might allow identification of those at increased risk of mortality and poor functional outcomes (Biffi et al. 2011).
2.6 Epilepsy
Epilepsy is mostly a multifactorial disorder, although familial forms occur and some epilepsy genes have been identified. Currently, a trial-and-error approach is employed to choose the most effective AED for a patient from numerous choices, but ~30 % of all patients are resistant to AED therapy, which can be partially attributed to the presence of polymorphisms of genes encoding enzymes involved in AED metabolism. Considerable information is being generated by advances in genomic technologies. Integration of these techniques with functional biology and bioinformatics will improve our understanding of the genetic contribution to epilepsy, the use of genetic testing for risk assessment, and personalized treatment (Kearney 2012). Personalized whole exome sequencing is already available, and whole genome sequencing is likely to be routinely available within the next few years.
One of the approaches to optimize AED therapy is pharmacogenomic testing to detect polymorphisms that may affect efficacy, tolerability, and safety of AEDs that include variations in the genes encoding drug-metabolizing enzymes such as cytochrome P450 or drug transporters such as MDR1 and MRP2 (Yoshida et al. 2011). The SNP association analysis shows that malic enzyme 2 (ME2) gene predisposes to idiopathic generalized epilepsy (Greenberg et al. 2005). ME2 is a genome-coded mitochondrial enzyme that converts malate to pyruvate and is involved in the neuronal synthesis of the neurotransmitter gamma amino butyric acid (GABA). The disruption of the synthesis of GABA predisposes to seizures, which are triggered when mutations at other genes are present. It is also becoming increasingly clear that SNPs play an integral role in the variability in both pharmacokinetics and pharmacodynamics of antiepileptic drugs (AEDs). Gene expression patterns of children on valproic acid monotherapy differ according to whether they have continuing seizures or remain free from seizures. This information can be used for personalizing AED therapy. The publication of the human genome and increasing sophisticated and powerful genetic tools offer new methods for screening drugs and predicting serious idiosyncratic side effects.
Available evidence suggests that genetic variants from ABCC2 transporter may be associated with an altered response to AEDs. Results of a meta-analysis literature indirectly suggest the possible role of the ABCC2 transporter at the blood-brain barrier in altered drug response in patients with epilepsy (Grover and Kukreti 2013). The authors suggest further studies in different ethnic groups to investigate the effects of the ABCC2 haplotypic variants and perform stratified analysis on the basis of different phenotypic covariates.
No AED treatment guidelines based on pharmacogenetic data are yet available. There is a need for, and an opportunity to establish, standards specific to the conduct of future AED studies to improve the management of epilepsy.
2.7 Migraine
The pathophysiology of migraine is not well understood, and although some gene mutations have been associated with special forms of migraine, genetic influences on common migraine at the population level were previously unknown. Genome-wide analysis of a large population in Europe, including migraineurs and non-migraineurs, revealed that two SNPs, rs2651899 and rs10166942, were associated with migraine, but the association was not preferential for migraine with aura or without aura, or with any specific features of migraine (Chasman et al. 2011).
2.8 Chronic Fatigue Syndrome
Chronic fatigue syndrome (CFS) is a complex illness that includes alterations in multiple body systems and results from the combined action of many genes and environmental factors. Genomic studies including SNPs have linked CFS to five mutations in three genes coding for the glucocorticoid receptor, for serotonin, and for tryptophan hydroxylase that are related to the body’s ability to handle stress (Vernon and Reeves 2006). The findings provide evidence of the biological basis of CFS and could lead to improved diagnostic tools and new therapies. Using an integrated genomic approach, a study suggests the possible role of genes involved in glutamatergic neurotransmission and circadian rhythm in CFS and supports further study of novel candidate genes in independent populations of CFS subjects (Smith et al. 2011). Another study used functional and structural equation modeling approaches to assess the contributions of the polymorphism (rs6311), DNA methylation, and clinical variables to HTR2A expression in CFS subjects from a population-based study (Falkenberg et al. 2011). Results suggest that rs6311 can affect both transcription factor binding and promoter methylation, and this along with an individual’s stress response can impact the rate of HTR2A transcription in a genotype- and methylation-dependent manner.
2.9 Role of Genomics in the Personalized Management of Psychiatric Disorders
The role of genomics and sequencing in psychiatric disorders was briefly described in Chap. 1 with limitations on the translation of this knowledge into clinical practice. Some examples of the applications of genomics in personalized management of psychiatric disorders are given in the following sections.
2.9.1 COMT Genotype and Response to Amphetamine
Catechol-O-methyltransferase (COMT) is an enzyme, a molecule responsible for breaking down dopamine and norepinephrine in parts of the brain. The activity of this enzyme is controlled in part by genetic factors. In certain individuals, COMT activity is higher than the average, which can lead to increased dopamine breakdown and therefore lower levels of dopamine in the frontal lobe. This may have behavioral consequences, such as difficulty with memory and concentration, as well as experiencing symptoms of depression. A functional polymorphism (val158-met) in the COMT gene has been shown to modulate prefrontal dopamine in animals and prefrontal cortical function in humans. COMT genotype has an effect on response to monoaminergic drugs.
Monoamines subserve many critical roles in the brain, and monoaminergic drugs such as amphetamine have a long history in the treatment of neuropsychiatric disorders and also as a substance of abuse. The clinical effects of amphetamine are quite variable, from positive effects on mood and cognition in some individuals to negative responses in others, perhaps related to individual variations in the monoaminergic function and monoamine system genes. Amphetamine enhanced the efficiency of prefrontal cortex function assayed with functional MRI during a working memory task in subjects with the high enzyme activity val/val genotype, who presumably have relatively less prefrontal synaptic dopamine, at all levels of task difficulty. In contrast, in subjects with the low activity met/met genotype who tend to have superior baseline prefrontal function, the drug had no effect on cortical efficiency at low-to-moderate working memory load and caused deterioration at high working memory load. These data illustrate an application of functional neuroimaging in pharmacogenomics and extend basic evidence of an inverted-U functional-response curve to increasing dopamine signaling in the prefrontal cortex. Further, individuals with the met/met COMT genotype appear to be at increased risk for an adverse response to amphetamine.
Related posts:
 RNA Sequencing from Laser Capture Microdissected Brain Tissue to Study Normal Aging and Alzheimer’s Disease
RNA Sequencing from Laser Capture Microdissected Brain Tissue to Study Normal Aging and Alzheimer’s Disease
 An Overview of Methods Used in Neurogenomics and Their Applications
An Overview of Methods Used in Neurogenomics and Their Applications
 Targeted Re-sequencing in Psychiatric Disorders
Targeted Re-sequencing in Psychiatric Disorders
 Location Analysis and Expression Profiling Using Next-Generation Sequencing for Research in Neurodegenerative Diseases
Location Analysis and Expression Profiling Using Next-Generation Sequencing for Research in Neurodegenerative Diseases
 Gene Expression-Based Approaches to Understanding Huntington’s Disease and New Tools for the Interpretation of Expression Datasets
Gene Expression-Based Approaches to Understanding Huntington’s Disease and New Tools for the Interpretation of Expression Datasets
 1: Narcolepsy—Unequivocal Diagnosis After Split-Screen, Video-Polysomnographic Analysis of a Prolonged Cataplectic Attack
1: Narcolepsy—Unequivocal Diagnosis After Split-Screen, Video-Polysomnographic Analysis of a Prolonged Cataplectic Attack
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


