Fig. 5.1
Histological changes in spinal cord tissue 48 h after the compression injury. Rats were treated with thoracic cord compression by a 20-g weight for 20 min (b, c) or laminectomy alone (a; sham). Compressed part of the cords were subjected to lectin staining (a, b) or OX-42 staining (c). Only a few lectin-positive cells were observed in the thoracic cord of the sham animals (a). On the other hand, lectin-positive cells (arrows) were remarkably increased in the compressed cords (b). OX-42-positive cells (arrows) were also observed in the compressed cord (c). (From [14] with permission)
The proliferated microglia (positive to OX42 staining) strongly expressed TNF-α and iNOS proteins (Fig. 5.2). These results suggest that microglia proliferated for several days after the spinal cord ischemic injury, and activated microglia modified neurodegenerative processes by secreting cytokines and nitric oxide. Microglia proliferation in the spinal cord was also observed in the model of peripheral nerve injury. Mika et al. reported that microglia proliferation was strongly observed in the dorsal lumbar spinal cord after peripheral nerve injury [16].
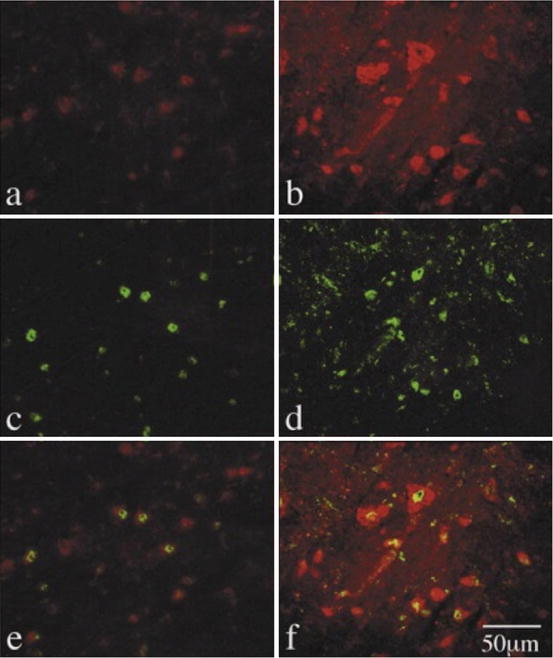
Fig. 5.2
Double staining of OX-42/TNF-α and OX-42/iNOS. Freezing sections were exposed to anti-TNF-α antibody (a, c, e) or anti-iNOS antibody (b, d, f) in the presence of anti-OX42 antibody. (a) TNF-α positive cells, (b) iNOS positive cells, (c) and (d) OX-42 positive cells, (e) double staining of OX-42/TNF-α, and (f) double staining of OX-42/iNOS. (From [14] with permission)
5.3 Detrimental Effects of Microglia in the Degeneration Processes of SCI
Although microglia may play an important role against infection in the CNS, overstimulation of this immune reaction may accelerate neuronal damage after SCI. Figure 5.3 shows a schema of the detrimental effects of microglia in the central and peripheral nervous systems.
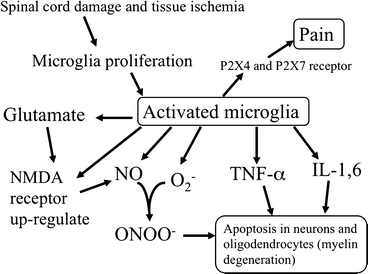
Fig. 5.3
Schema of the detrimental effects of activated microglia after injury of the nervous system
Pathologically activated microglial cells release various harmful components such as nitric oxide (NO), superoxide (O2 −), and several kinds of cytokines including interleukin (IL)-1 and IL-6 and tumor necrosis factor-α (TNF-α), which can directly or indirectly affect biological functions of neurons, aggravate neurotoxicity, and may even eventuate downstream in exacerbating neurodegeneration and the pathway of cell death [17–22]. Zhang and Fedoroff [23] reported that neurons in mixed neuron-microglia cultures survived longer than those in pure neuron cultures; however, when they treated the mixed culture by lipopolysaccharide (LPS), microglia impeded neuronal survival. These papers indicate that substances (cytokines and nitric oxide) released from activated microglia is possibly toxic to neurons. Neumann et al. [24] reported that application of recombinant TNF inhibited neurite outgrowth and branching of cultured hippocampal neurons. This effect was absent in the neurons of TNF receptor-deficient animals. Westmoreland et al. [25] reported that the addition of TNF-α induced cell death in cultured human neuronal cell lines and pentoxifylline, which inhibits the effects of TNF-α, had a significant protective effect in this assay. Viviani et al. [26] reported that activation of TNF-α release from glial cells induced neuronal death, which was inhibited by the addition of TNF-α antibody in a glia-neuron co-culture. Other cytokines, released from activated microglia, also influence the progress of secondary neuronal damage. Interlrukin-1 (IL-1) has been reported to induce neuronal damage in both acute and chronic neurodegenerative disease [27] and cell death caused by ischemia, brain injury, or excitotoxins [28]. Interleukin 6 (IL-6) has also been reported to be toxic to neurons. Chronic exposure to IL-6 during neuronal development induced cell damage and death in a subpopulation of developing granule neurons, and increased the susceptibility of the granule neurons to a toxic insult produced by excessive activation of NMDA receptors [29]. In addition, IL-6-deficient mice showed an increased resistance to glutamate toxicity [30].
Nitric oxide (NO) and superoxide (O2 −), which are released by activated microglia, are also reported to be toxic to the neuronal system due to production of peroxynitrite (ONOO−) during degeneration [31]. There is evidence that the concomitant production of superoxide, required for the generation of peroxynitrite by NO, is provided by pathologically activated microglia [32]. The particularly high neurotoxicity of ONOO−, which destroys neuronal cell membranes, has been shown in previous studies [33, 34].
The possible contribution of microglia by glutamate signaling has been reported. The activation of ionotropic glutamate receptors in microglia has deleterious consequences to neurons and oligodendrocytes. NMDA receptor expression was upregulated in activated microglia following ischemia [35, 36]. The activation of NMDA receptors in microglia leads to NO release. Pharmacological blockade of NMDA receptors and inhibition of iNOS prevents oligodendrocyte cell death [37]. Thus, NMDA receptor blockade protects oligodendrocytes by reducing the release of NO from microglia [37, 38]. Activated microglia itself can release glutamate which directly induces damage of neurons and oligodendrocytes. Both resting and activated microglia release glutamate through the cystine/glutamate antiporter [39]. However, glutamate released through resting microglia is rapidly and efficiently removed by glutamate transporters in other glial cells, mainly astrocytes. In contrast, the activation of microglia induces the release of factors, such as reactive oxygen species, TNF-α, and IL-1β that impair the function of glutamate transporters, resulting in an increase in extracellular levels of glutamate [39].
Peripheral nerve injury (neuropathic pain) and peripheral tissue injury (inflammation) induce abnormal pain sensation such as hyperalgesia and allodynia. Central sensitization, which is an enhanced responsiveness of nociceptive neurons in the CNS, is thought to be a mechanism for hyperalgesia and allodynia. Recent studies revealed that microglia in the spinal dorsal horn are activated in response to peripheral nerve or tissue injury and are involved in persistent pain sensation [40–43]. Inhibition of microglia proliferation and activation in spinal cord with minocycline [16, 44, 45] and propentofylline [46] ameliorated allodynia and hyperalgesia after peripheral nerve injuries. These results strongly indicate that the activation of microglia directly induced neuropathic and inflammatory pain.
The key mechanism of microglia-related pain is mediated via purinergic ionotropic receptors, such as P2X4 and P2X7, in activated microglia [47]. Tsuda et al. [48] reported that P2X4 receptor expression increased strikingly in the ipsilateral spinal cord after peripheral nerve injury, and P2X4 receptors were induced in hyperactive microglia but not in neurons or astrocytes. Pharmacological blockade of spinal P2X4 receptors reversed tactile allodynia caused by peripheral nerve injury without affecting acute pain behaviors in naive animals. Kobayashi et al. [49] reported that both P2X7 mRNA and protein increased in the spinal cord after peripheral nerve injury. Double-labeling studies revealed that cells expressing increased P2X7 mRNA and protein after nerve injury were predominantly microglia in the dorsal horn. Intrathecal application of a P2X7 antagonist suppressed mechanical hypersensitivity after the peripheral injury.
5.4 Beneficial Effects of Microglia After SCI
The effects of activated microglia on SCI are not limited to the harmful side. Activated astrocytes and microglial cells also confer beneficial effects on injured neurons and interfere with neurogenesis [50–52]. Prewit et al. [53] reported that increasing the presence of activated macrophage/microglial cells at a SCI site can provide an environment beneficial to the promotion of regeneration of sensory axons. Rabchevsky et al. [54] reported that implantation of cultured microglia promote neural regeneration after SCI.
Well-controlled inflammation, depending on the timing and amount, may possibly lessen and/or repair CNS damage. Nakanishi et al. [55] reported that cultured microglia released leukemia inhibitory factor (LIF), which promotes the differentiation of neural stem/progenitor cells. Kerr and Patterson [56] showed that LIF over-expression also causes the development of severe hindlimb motor dysfunction, an effect mediated by the enhanced activation of microglia/macrophages. To the contrary, they also reported that daily administration of LIF (25 mg/kg/day) promotes oligodendrocyte survival after SCI [57].
Growing evidence showed that microglial cells, under certain circumstances, can exert neuroprotective and supportive effects and enhance synthesis of neurotrophic factors.
Kerr and Patterson [57] reported that insulin-like growth factor-1(IGF-1) released from microglia promoted neuronal regeneration. Lehrmann et al. [58] reported that microglia is a major source of transforming growth factor (TGF)-β1 production after CNS injury. Application of TGF-β1 decreased infarct size after focal cerebral ischemia [59] and protected hippocampal neurons after global cerebral ischemia [60].
Nerve growth factor (NGF), a typical neuroregenerative protein, has also been reported to release from microglia in several CNS damage conditions [61, 62]. Other neuroprotective factors, such as brain-derived neurotrophic factor (BDNF) and glial cell line-derived neurotrophic factor (GDNF), have been reported to release from microglia in some pathological conditions of the CNS [63, 64]. Yang et al. [65] reported that mechanically injured astrocyte-conditioned medium (ACM) could provoke beneficial activation of microglial cells. Microglial cells released BDNF through the exposure of ACM, and the neurons cocultured with microglia showed the promotion of neurite outgrowth, which was suppressed by the addition of BDNF neutralizing antibody. Specific inhibition of the p38 mitogen-activated protein kinase inhibited the effects of ACM, including BDNF promotion and subsequent neurite outgrowth.
As mentioned above, microglia also contribute to the regeneration and preservation of damaged neurons and oligodendrocytes via the releasing of some trophic substances. However, it is still uncertain what causes the beneficial activation of microglial cells after CNS injury.
5.5 Possible Therapeutic Modules by Modification of Microglial Function for the Treatment of SCI
To summarize so far, microglia have two faces on the processes of neuronal degeneration after SCI. It has been suggested that the level of the inflammatory stimulus regulates the level of the microglial response. For example, Li et al. [66] demonstrated that, although high levels of LPS (>1 μg/mL) induced proinflammatory cytokine expression and neurotoxicity, lower concentrations (<500 ng/mL) increased neuronal cell viability and promoted neurite extension.
In recent years, a new classification of microglia has entered the literature. Described as an alternatively activated subset of microglia (M2), these cells have markers that can differentiate them from classically activated microglia (M1) [67, 68]. The M2 microglia are typically considered to be less inflammatory than M1 microglia; they are characterized by reduced NO production and increased anti-inflammatory cytokine production. The M2 microglia are involved in tissue repair, wound healing, and extracellular matrix remodeling [67]. After injury or in vitro following exposure to IL-4 or IL-13, microglia develop this nontoxic M2 phenotype, resulting in extensive neurite elongation and outgrowth across inhibitory surfaces [68, 69]. Therefore, control of microglia function may become a novel therapeutic strategy for the treatment of SCI.
The inhibition of pathological immune reactions by classically activated microglia (M1) may ameliorate secondary neurodegeneration in the acute stage after SCI. Several substances have been reported to inhibit microglial proliferation or functions (Table 5.1).
Table 5.1
Candidates of therapeutic modalities for SCI by inhibition of microglial reaction
Year | Authors | Candidates (substances or treatments) | Experimental methods | Findings | Reference no. |
|---|---|---|---|---|---|
1996 | Ogata and Schubert | Adenosine receptor analog | Microglia culture | Induction of microglia apoptosis | [70] |
1997 | Nakamura et al. | Adenosine receptor analog propentofylline | Microglia culture | Inhibition of microglial proliferation and phagocytosis ability | [71] |
1998 | Si et al. | Propentofylline cAMP analog | Microglia culture | Inhibition of microglia proliferation and release of cytokines, NO, and O2 − | [21] |
2002 | Chuai et al. | Prostaglandin E1 analog | Microglia culture | Inhibition of TNF-α and NO release | [77] |
2003 | Ogata et al. | ATP and selective P2y-receptor analog | Microglia culture | Inhibition of cytokines (TNF-α, IL-1β, IL-6) and NO release | [80] |
2007 | Yune et al. | Minocycline | SCI contusion model | Improvement of motor function and inhibition of pro-nerve growth factor production in microglia | [72] |
2007 | Tawfik et al. | Propentofylline | Peripheral nerve injury | Inhibition of microglia proliferation and induction of pain relief | [46] |
2008 | Morino et al. | Systemic hypothermia | SCI compression model | Improvement of motor function and inhibition of microglia proliferation | [86] |
2009 | Mika et al. | Minocycline, pentoxifylline | Peripheral nerve injury | Inhibition of microglia proliferation and induction of pain relief | [16] |
2012 | Morizane et al. | Local hypothermia | SCI contusion model | Improvement of motor function and inhibition of microglia proliferation and TNF-α production | [87] |
Microglia proliferation/activation can be controlled by some substances. Cl-adenosine, an unselective agonist of adenosine receptors, induced apoptosis of rat-cultured microglia [70]. Nakamura et al. [71] reported that an adenosine receptor analog and propentofylline, a xanthine derivative, inhibited microglia proliferation and phagocytosis activity of microglia. Minocycline, a second-generation tetracycline, has been reported to decrease microglia/macrophage activation and proliferation [72–74]. Application of minocycline reduced delayed oligodendrocyte death, attenuated axonal degeneration, and improved functional outcome after SCI [75, 76]. Tawfik et al. [46] reported that propentofylline treatment to nerve injured rats resulted in significantly decreased microglia proliferation in the spinal cord tissue and reversal of nerve injury-induced allodynia. Mika et al. [16] also found that chronic intraperitoneal injection of minocycline and pentoxifylline (cytokine inhibitor) inhibited activation of microglia and diminished neuropathic pain symptoms after peripheral nerve injury.
Pharmacological control of toxic substances release from microglia has been attempted using cultured microglia. Si et al. [21] reported that propentofylline inhibited cytokine release from cultured microglia by cAMP elevation. Adenosine induces adenylate cyclase activation via adenosine receptors, which provides intracellular cAMP elevation. Propentofylline increases cAMP as phosphodiesterase inhibition. Thus, the inhibitory effect of adenosine and propentofylline on microglia function is mediated by a common intracellular mechanism, such as cAMP-dependent kinase.
We have reported that alprostadil alfadex, a prostaglandin E1 (PGE1) analog, showed inhibition of LPS-induced release of TNF-α and NO from rat-cultured spinal cord microglia [77]. The inhibitory effects of the PGE1 analog may be mediated by EP1 and EP2 receptor subtypes. Caggiano and Kraig [78] demonstrated that EP1 and EP2 receptor subtypes were present in rat-cultured microglia. The EP1 receptor is related to intracellular calcium elevation, and adenylate cyclase activity is increased by EP2 receptor stimulation [79]. In our data [77], inhibition of protein kinase A by H89 inhibited the PGE1 action on LPS-induced TNF-α release, but showed no effect on NO release. The working mechanism of PGE1 on TNF-α release may involve the EP2 receptor-adenylate cyclase-cyclic AMP-protein kinase A pathway. On the other hand, the protein kinase C inhibition by staurosporine blocked the PGE1 action on LPS-induced NO release. These results suggest that PGE1 inhibited the TNF-α (cytokine) release and NO release by different mechanisms, such as activation of protein kinase A and protein kinase C.
Adenosine triphosphate (ATP) is also a strong inhibitor of microglia action [80]. We have tested the effects of endogenous purines (ATP, ADP, CTP, UDP, UTP) and P2y receptor agonists (ADPβS and 2-methylthio-ATP) on LPS-induced cytokine (TNF-α, IL-1β and IL-6) release from cultured spinal cord microglia. The rank order of inhibitory potency of endogenous purines on TNF-α release was: ATP > ADP >> UTP > UDP > CTP. The addition of 10 μM 2-methylthio-ATP showed a similar potency as 100 μM ATP. The addition of ADPβS, however, showed less effect. These endogenous purines and selective ATP receptor agonists showed a similar inhibitory effect in their rank order on LPS-induced IL-6 release. The application of P2y receptor agonists might be considered as a pharmacological treatment for several pathological conditions of the spinal cord, including toxic immunoreactions.
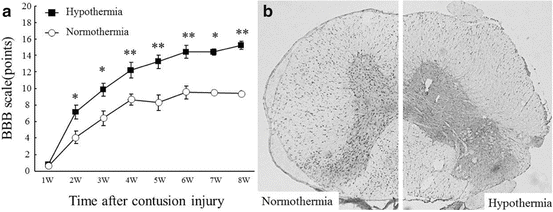
Cooling the damaged tissue should be a powerful module on inflammatory responses after SCI. Systemic cooling has been reported to protect neurons after traumatic or ischemic brain damage [81, 82]. Using several SCI models, systemic hypothermic treatment under general anesthesia also showed successful neuroprotection after CNS injury [83–85]. The target of hypothermic treatments may be, at least in part, somewhat delayed in response after injury. Using the thoracic (T11) spinal cord mild compression model [14], we performed systemic mild hypothermia treatment (33 °C) under general anesthesia for 1 h after the injury [86]. Compared with normothermic animals (37 °C), remarkable inhibition of microglia proliferation and tissue TNF-α content were observed in parallel with recovery of motor activity in the hindlimbs. However, longer hypothermic treatment should be necessary for more severe injuries, such as the spinal cord contusion model. In order to achieve more stable and effective spinal cord cooling for the treatment of severe SCI, we developed a novel thermoelectric cooling device using Peltier modules [87]. Using this device, we performed local mild hypothermia (33 °C) for 48 h. During the hypothermic treatment, rats were awake and could move in the cage. Hindlimb motor function, evaluated using a BBB scale [88], in the hypothermic animals was significantly higher than that in the normothermic animals from 2 to 8 weeks after the injury (Fig. 5.4a). Compared with normothermic animals, the hypothermic treatment significantly reduced the number of OX42-positive cells (microglia; Fig. 5.4b) and TNF-α-positive cells. The TNF-α content in the injured spinal cord tissue was also significantly inhibited by hypothermic treatment. These results suggest that inhibition of microglia proliferation and activation by hypothermia is a reasonable therapeutic module for the treatment of acute SCI.
 Morphological Changes in Anterior Horn Cells, Immunoreactivity to Neurotrophic Factors, and Neuronal Cell Death of Spinal Cord Lesions in the Spinal Hyperostotic Mouse (twy/twy) with Chronic Mechanical Cord Compression
Morphological Changes in Anterior Horn Cells, Immunoreactivity to Neurotrophic Factors, and Neuronal Cell Death of Spinal Cord Lesions in the Spinal Hyperostotic Mouse (twy/twy) with Chronic Mechanical Cord Compression
 Visualization of Electrophysiological Activity in the Spinal Cord Using Magnetospinography
Visualization of Electrophysiological Activity in the Spinal Cord Using Magnetospinography
 Blockade of Interleukin-6 Effects on Cytokine Profiles and Macrophage Activation After Spinal Cord Injury in Mice
Blockade of Interleukin-6 Effects on Cytokine Profiles and Macrophage Activation After Spinal Cord Injury in Mice
 Recombinant Human Hepatocyte Growth Factor Promotes Functional Recovery After Spinal Cord Injury
Recombinant Human Hepatocyte Growth Factor Promotes Functional Recovery After Spinal Cord Injury
 Spinal Kyphosis Causes Demyelination and Neuronal Loss in the Spinal Cord
Spinal Kyphosis Causes Demyelination and Neuronal Loss in the Spinal Cord
 Oxidative Stress as Secondary Injury Mechanism After Mechanical Trauma in the Spinal Cord
Oxidative Stress as Secondary Injury Mechanism After Mechanical Trauma in the Spinal Cord




Related posts:
Morphological Changes in Anterior Horn Cells, Immunoreactivity to Neurotrophic Factors, and Neuronal Cell Death of Spinal Cord Lesions in the Spinal Hyperostotic Mouse (twy/twy) with Chronic Mechanical Cord Compression
Visualization of Electrophysiological Activity in the Spinal Cord Using Magnetospinography
Blockade of Interleukin-6 Effects on Cytokine Profiles and Macrophage Activation After Spinal Cord Injury in Mice
Recombinant Human Hepatocyte Growth Factor Promotes Functional Recovery After Spinal Cord Injury
Spinal Kyphosis Causes Demyelination and Neuronal Loss in the Spinal Cord
Oxidative Stress as Secondary Injury Mechanism After Mechanical Trauma in the Spinal Cord
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
