Fig. 1
Sleep state abnormalities in orexin receptor knockout mice. Representative 12-h dark-period (20:00–08:00) hypnograms for wild-type (WT), OX1R −/−, OX2R −/−, and OX1R −/−; OX2R −/− mice, all with a C57BL/6J background, are shown. The different levels above the baseline indicate states of sleep and wakefulness (e.g., REM, NREM, and wakefulness) of mice at the time. Episodes of direct transition from wakefulness to REM sleep are shown by arrows. Note the greater awake and NREM sleep episode fragmentation and reduced duration of wakefulness in the hypnograms of OX2R −/− and OX1R −/−; OX2R −/− mice compared with WT and OX1R −/− mice. Episodes of direct transition from wakefulness to REM sleep were not observed in OX1R −/− mice, and were hardly observed in OX2R −/− mice, though they were frequently observed in OX1R −/− and OX2R −/− mice. Modified from Sakurai (2007)
Collectively, mouse reverse genetic studies suggest that the normal regulation of wakefulness and NREM sleep transitions depends critically on OX2R activation, whereas the profound dysregulation of REM sleep control unique to narcolepsy emerges from loss of signaling through both OX1R- and OX2R-dependent pathways.
This conclusion has been further confirmed by a complementary experiment, i.e., comparing the arousal effects of ICV orexin-A administration between wild-type, OX1R −/−, and OX2R −/− mice (Fig. 2) (Mieda et al. 2011). The effects of orexin-A on wakefulness and NREM sleep were significantly attenuated in both OX1R −/− and OX2R −/− mice as compared to wild-type mice, with substantially larger attenuation in OX2R −/− than in OX1R −/− mice, suggesting the pivotal role of OX2R and the additional role of OX1R in the promotion of wakefulness. By contrast, the suppression of REM sleep via orexin-A administration was marginally and similarly attenuated in both OX1R −/− and OX2R −/− mice, suggesting a comparable contribution of the two receptors to REM sleep suppression (Mieda et al. 2011). The supplementary role of OX1R in the suppression of NREM sleep is consistent with the fact that OX2R −/− mice with a C57BL/6J, but not C57BL/6J-129/SvEv-mixed, genetic background show less fragmented wakefulness than orexin −/− mice and OX1R −/−; OX2R −/− mice (Hasegawa et al. 2014; Mochizuki et al. 2011; Sakurai 2007; Willie et al. 2003), which suggests that OX1R is indispensable for the maintenance of wakefulness in the absence of OX2R.
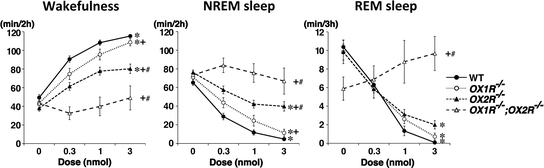
Fig. 2
Differential effects on sleep/wakefulness states following activation of OX1R and OX2R. Dose responses of the effect of ICV orexin-A in wild-type (WT), OX1R −/−, OX2R −/−, and OX1R −/−; OX2R −/− mice on time spent in wakefulness, NREM sleep, and REM sleep following administration. Modified from Mieda et al. (2011)
The conclusion drawn from mouse genetics apparently contradicts the fact that an inherited canine model of narcolepsy, which demonstrates a frequent occurrence of cataplexy as well as excessive sleepiness, is attributable solely to mutations of the OX2R gene (Lin et al. 1999). Species differences (e.g., the precise expression patterns of the two orexin receptors) and/or selection bias may explain such an inconsistency. It should be noted that, even in canines, the absence of orexin peptides may cause severe narcoleptic symptoms as compared to OX2R mutation. Early studies reported that narcoleptic Dobermans and Labradors with OX2R mutations were much less severely affected with cataplexy than poodles with sporadic narcolepsy, which were supposed to lack orexin peptides (Baker et al. 1982).
4 Sites of Expression of Orexin Receptors Relevant to the Physiological Control of Sleep/Wakefulness
The application of exogenous orexins has been shown to excite many types of neurons. Monoaminergic and cholinergic nuclei of the hypothalamus and brainstem involved in the regulation of sleep and wakefulness especially receive dense projections of orexin neurons, express orexin receptors, and are activated by the application of orexin peptides in slice preparations (Bayer et al. 2001; Brown et al. 2001; Burlet et al. 2002; Eggermann et al. 2001; Eriksson et al. 2001; Horvath et al. 1999; Liu et al. 2002; van den Pol et al. 2002; Yamanaka et al. 2002; Mieda et al. 2013). Furthermore, the administration of orexin-A directly into the LC (Bourgin et al. 2000), TMN (Huang et al. 2001), BF cholinergic area (Espana et al. 2001; Thakkar et al. 2001), and LDT (Xi et al. 2001) has also been reported to increase wakefulness. However, neurons activated by the pharmacological application of exogenous orexin may not necessarily be essential to the endogenous mechanisms by which orexin neurons regulate sleep and wakefulness in a physiological condition. Thus, neurons directly downstream from orexin neurons in physiological conditions (i.e., the site and subtype of orexin receptors that mediate the wake-promoting and REM-gating effects by endogenous orexins) have remained incompletely understood.
Histaminergic neurons in the TMN, which express OX2R exclusively, are good candidates for such downstream neurons contributing the arousal effect of orexin. Wake-promoting effect of ICV orexin-A administration is both markedly attenuated by the histamine H1 receptor (H1R) antagonist pyrilamine (Yamanaka et al. 2002) and is absent in H1R −/− mice (Huang et al. 2001). In addition, Mochizuki et al. (2011) produced OX2R-deficient mice by inserting a loxP-flanked transcription-disrupter (TD) gene cassette into the OX2R gene, in which normal OX2R expression could be restored by Cre recombinase-mediated excision of TD cassette. Using such an elegant genetic model, they showed that focal restoration of OX2R expression in the TMN and adjacent regions completely reversed the fragmentation of wakefulness episodes observed in their OX2R-deficient mice.
However, this hypothesis remains controversial. Mice lacking both OX1R and H1R demonstrate no abnormality in sleep or wakefulness, which contradicts the idea that H1R-mediated histaminergic pathway is the principal downstream component of OX2R-mediated orexinergic signaling (Hondo et al. 2010). Moreover, a recent study showed that increased probability of sleep-to-wakefulness transitions by optogenetic activation of orexin neurons does not depend on histamine (Carter et al. 2009).
Recently, we searched for monoaminergic and cholinergic nuclei of the brainstem and hypothalamus in which the focal restoration of orexin receptor expression by recombinant AAV vectors ameliorates narcoleptic phenotype of OX1R −/−; OX2R −/− mice (Fig. 3a) (Hasegawa et al. 2014). If the regional restoration of orexin receptors in a certain brain region suppresses narcoleptic symptoms in these mice, that particular region can at least be regarded as one of the important downstream targets of orexin neurons. The targeted restoration of orexin receptor expression in the DR and LC of these mice differentially inhibited cataplexy-like episodes and the fragmentation of wakefulness (i.e., sleepiness), respectively. The suppression of cataplexy-like episodes correlated with the number of serotonergic neurons restored with orexin receptor expression in the DR, while the consolidation of fragmented wakefulness correlated with the number of noradrenergic neurons restored in the LC. Furthermore, the pharmacogenetic activation of these neurons using DREADD (designer receptor exclusively activated by designer drug) technology ameliorated narcolepsy in mice that lacked orexin neurons (Fig. 3b). These results suggest that DR serotonergic and LC noradrenergic neurons play differential roles in the regulation of sleep and wakefulness by orexin neurons.
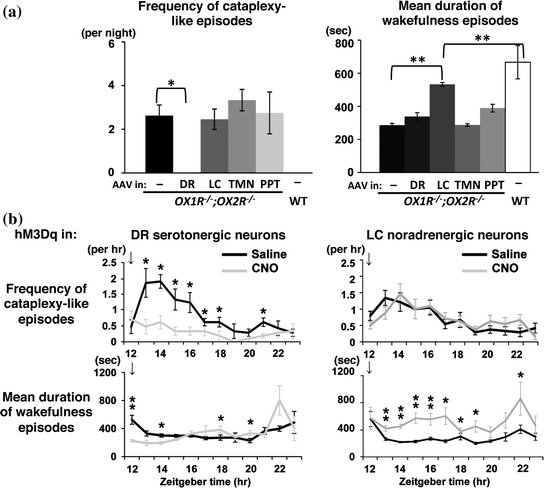
Fig. 3
Search for the downstream targets of orexin neurons to prevent narcolepsy. a Restoration of orexin receptor expression in the DR and LC inhibits cataplexy-like episodes and consolidates wakefulness, respectively, in narcoleptic OX1R −/−; OX2R −/− mice. Orexin receptor expression was restored in the nuclei indicated in OX1R −/−; OX2R −/− mice by recombinant AAV vectors. b Pharmacogenetic activation of DR serotonergic and LC noradrenergic neurons suppresses cataplexy-like episodes and consolidates wakefulness, respectively, in narcoleptic mice lacking orexin neurons (orexin/ataxin–3 mice). Orexin/ataxin–3 mice with DR serotonergic neuron–selective or LC noradrenergic neuron–selective expression of excitatory DREADD hM3Dq were injected with saline or CNO, the artificial ligand for DREADDs. Hourly plots of number of cataplexy-like episodes and mean duration of wakefulness episodes within 12 h after saline or CNO administration at ZT 12 (arrows) are shown. Modified from Hasegawa et al. (2014)
The suppression of cataplexy-like episodes by DR serotonergic neurons, but not by LC noradrenergic neurons, was an unexpected result (Hasegawa et al. 2014), because previous pharmacological and electrophysiological studies suggested that LC noradrenergic neurons are good candidates for downstream neurons to prevent cataplexy. For example, drugs that increase noradrenergic tone strongly suppress cataplexy in humans and canines, while blocking the noradrenergic signaling increases the frequency of cataplexy (Hirai and Nishino 2011; Nishino and Mignot 1997). In addition, LC neurons cease firing during cataplexy in canines (Wu et al. 1999). Nevertheless, our observations never deny the clinical importance of enhancing noradrenergic systems for preventing cataplexy, yet simply indicate that the sole regulation of LC noradrenergic neurons by endogenous orexins is not sufficient to suppress cataplexy in narcoleptic mice. Non-LC noradrenergic neurons may also play an important role in the suppression of cataplexy by the pharmacological augmentation of systemic noradrenergic tone, which may be independent of the orexinergic regulation.
The contribution of orexin signaling in DR serotonergic neurons in the suppression of cataplexy fits with the observations that these neurons express both OX1R and OX2R (Mieda et al. 2011) and that the disruption of both OX1R- and OX2R-mediated pathways is required for the frequent occurrence of cataplexy, as described earlier (Sakurai 2007) (Fig. 4a). DR serotonergic neurons greatly reduce firing rates during cataplexy in canines (Wu et al. 2004). In addition, these neurons, as well as LC noradrenergic neurons, have been implicated in the suppression of REM sleep by inhibiting REM-on cholinergic neurons in the PPT/LDT and/or by activating REM-off GABAergic neurons in the ventrolateral periaqueductal gray (vlPAG) and adjacent lateral pontine tegmentum (LPT), also known as dorsal deep mesencephalic reticular nuclei (dDpMe) (Pace-Schott and Hobson 2002; Luppi et al. 2011). Indeed, we observed dense projections of DR serotonergic neurons to these brain areas, as well as to the amygdala (Hasegawa et al. 2014), which suggests that DR serotonergic neurons may coordinately control multiple brain regions involved in the regulation of REM sleep and emotion.
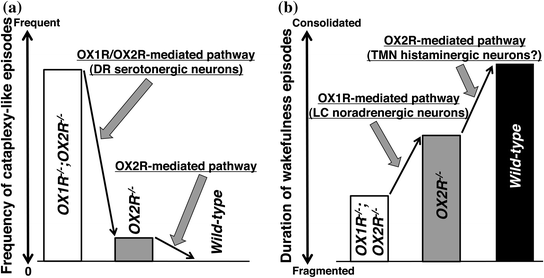
Fig. 4
Proposed model for OX1R- and OX2R-mediated pathways in suppressing narcoleptic symptoms. a Prevention of cataplexy-like episodes. b Consolidation of wakefulness episodes. Modified from Hasegawa et al. (2014)
As described above, the fragmentation of wakefulness is less severe in OX2R −/− mice than in orexin −/− mice and OX1R −/−; OX2R −/− mice with C57BL/6J genetic background (Mochizuki et al. 2011; Sakurai 2007), suggesting that OX1R plays an important role in the maintenance of wakefulness in the absence of OX2R (Mieda et al. 2011). Indeed, restoration of OX1R expression in the LC noradrenergic neurons of OX1R −/−; OX2R −/− mice stabilized wakefulness episodes to an extent comparable to those in OX2R −/− mice (Hasegawa et al. 2014). Considering the fact that LC noradrenergic neurons exclusively express OX1R in wild-type mice (Mieda et al. 2011), these neurons may be responsible for the contribution of OX1R to the maintenance of wakefulness, while another OX2R-mediated mechanism, most likely mediated by TMN histaminergic neurons, is further required for fully maintained wakefulness as in normal mice (Fig. 4b). Recent optogenetic studies have provided supports for the importance of the orexinergic regulation of LC noradrenergic neurons in the consolidation of wakefulness. For instance, there is a causal relationship between the firing of LC noradrenergic neurons and transitions from sleep to wakefulness (Carter et al. 2010). Moreover, the optogenetic inactivation of these neurons prevents the arousal effects of the optogenetic stimulation of orexin neurons (Carter et al. 2012).
5 Pharmacological Dissection of Sleep/Wakefulness Regulation by Orexin Receptors
A series of non-selective (dual) antagonists for orexin receptors (DORA), as well as subtype-selective antagonists (SORA), have been developed. On the one hand, these drugs are drawing people’s attention as novel medications for insomnia and other diseases (Scammell and Winrow 2011; Sakurai 2014). On the other hand, they are also useful for studying the roles of each subtype in the regulation of sleep/wakefulness.
To a large extent, results obtained by pharmacological studies utilizing DORAs and SORAs are consistent with those derived from genetic studies described in the previous sections. Selective blockade of OX2R efficiently increases NREM sleep and shortens NREM sleep latency (Dugovic et al. 2009, 2014; Betschart et al. 2013; Etori et al. 2014; Gozzi et al. 2011). Blockade of both OX1R and OX2R does increases NREM sleep, but also causes disproportionally large increase in REM sleep (Bonaventure et al. 2015; Dugovic et al. 2009, 2014; Etori et al. 2014; Hoyer et al. 2013). Except one study showing increases in REM and NREM sleep with SB-334867 (Morairty et al. 2012), selective blockade of OX1R alone does not cause any effects with statistical significance on baseline sleep/wakefulness (Bonaventure et al. 2015; Dugovic et al. 2009, 2014; Gozzi et al. 2011; Steiner et al. 2013). Thus, OX2R is the principal regulator of wakefulness/NREM sleep transition, while both OX1R- and OX2R-mediated pathways are critical for gating REM sleep.
Administration of DORAs seldom induces cataplexy in normal animals, although there is a report that less than half of rats treated with high doses of SB-649868 demonstrated direct transitions from wakefulness to REM sleep (Dugovic et al. 2014). Therefore, as compared to the induction of NREM and REM sleep, nearly complete and/or chronic absence of both OX1R- and OX2R-mediated pathways may be needed for cataplexy to occur. This notion is consistent with the observation that degeneration of more than 95 % of orexin neurons is required for the occurrence of cataplexy in mice, whose frequency subsequently increases along with further degeneration (Tabuchi et al. 2014).
Related posts:
 Hypocretin Story
Hypocretin Story
 Models of Narcolepsy
Models of Narcolepsy
 Induced Modulation of REM Sleep and Its Loss Associated Patho-Physiological Changes Are Mediated Through Locus Coeruleus
Induced Modulation of REM Sleep and Its Loss Associated Patho-Physiological Changes Are Mediated Through Locus Coeruleus
 and Circadian Influences in Sleep and Psychiatric Disorders: A Review of Experimental and Computational Modelling Studies
and Circadian Influences in Sleep and Psychiatric Disorders: A Review of Experimental and Computational Modelling Studies
 of Neuronal Circuitry Involved in the Regulation of Sleep/Wakefulness Using Optogenetics
of Neuronal Circuitry Involved in the Regulation of Sleep/Wakefulness Using Optogenetics
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


