Insult
Effect
Axonal degeneration
Neuronal apoptosis
References
6OHDA
Some axons preserved for ~11 days
+
−
Sajadi et al. (2004)
MPTP
Enhances survival, prevents nigrostriatal axon degeneration, and attenuates neurotransmitter loss
+
−
Hasbani and O’Malley (2006)
Taxol
Resistant to paclitaxel neuropathy by behavioral, electrophysiological, and pathological measures.
+
ND
Wang et al. (2002)
Diabetes
Alleviated abnormal sensory responses, nerve conduction, retina dysfunction and reduction of surviving retinal ganglion cells
+
+
Zhu et al. (2011)
P0 null
Reduces motoneuron loss and axonal loss at ~3 months
+
+
Samsam et al. (2003)
PMP22 null
Reduces axonal loss and clinical impairments without altering demyelination
+
ND
Meyer zu Horste et al. (2011)
SOD1 mutant transgene (G37R, G85R, and G93A)
Modest exention in lifespan (G93A)
−
−
pmn
Attenuates symptoms, extends life span, prevents axonnal degeneration, rescues motoneuron number and size, and delays retrograde transport deficits
+
+
Ferri et al. (2003)
gad
Reduction of pathology at 4 month
+
ND
Mi et al. (2005)
sma
No protective effect
−
−
8.2 Wallerian Degeneration
Axonal degeneration is observed in a wide variety of pathological conditions. A classic example of axonal degeneration is Wallerian degeneration, which occurs after an axon is cut (Waller 1851). Wallerian degeneration is observed as a major component in many neurodegenerative diseases, such as Parkinson’s Disease and Alzheimer’s Disease (Fig. 8.1a) (Coleman 2005; Saxena and Caroni 2007; Wang et al. 2012). An active and evolutionarily conserved mechanism drives Wallerian degeneration (Fang and Bonini 2012). Wallerian degeneration can be investigated in both the peripheral and central nervous system (PNS and CNS) using injury-induced axonal degeneration models in vivo and in vitro (Coleman and Freeman 2010; Wang et al. 2012). Acutely severed axons exhibit the profiles of microtubule disassembly, blebbing of the distal axon, followed by axonal fragmentation; similar processes of Wallerian degeneration have been observed in several organisms including fruit flies and nematodes (Coleman and Freeman 2010). Some chemically induced acute injuries such as the local destruction of microtubule structure, with the microtubule disrupting anti-cancer agent vinblastine, induce axonal degeneration closely resembling Wallerian degeneration, and should, therefore, be considered as equivalent to physical lesions (Fig. 8.1a). Thus, understanding the mechanisms of axonal degeneration in simple traumatic injuries allows us to model how axons are lost in more complex neurodegenerative conditions.
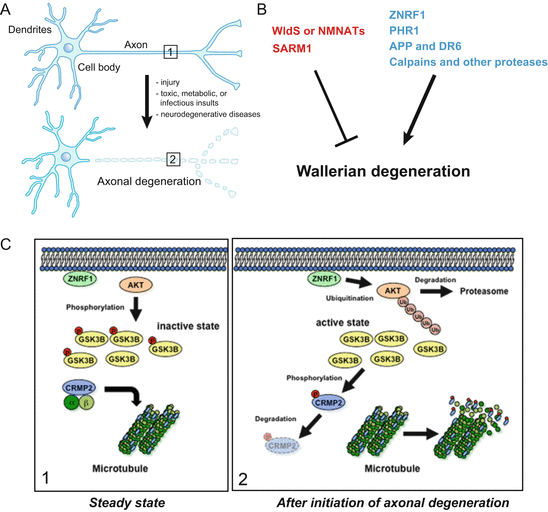
Fig. 8.1
Molecular pathways regulating Wallerian degeneration. (a) When an axon is cut, the isolated distal portion of axon rapidly undergoes Wallerian degeneration. Similar degeneration is also observed in the nerves in many neurological disorders including neurodegenerative diseases. (b) Molecules that mediate axonal degeneration are the E3 ubiquitin ligase ZNRF1, PHR1, or the Ca2+-activated cysteine protease Calpains. Loss of these molecules can protect axons from degeneration. By contrast, wlds and NMNATs can inhibit the progression of axonal degeneration (see text for details). (c) The ZNRF1–AKT–GSK3B–CRMP2 pathway controls cytoskeletal integrity during axonal degeneration. In the steady state axon (boxed area 1 in a, left panel), AKT phosphorylates GSK3B and thereby inactivates it. CRMP2 is not phosphorylated in the steady state axons and continues to maintain the microtubule architecture. Upon initiation of axonal degeneration (boxed area 2 in a, right panel), ZNRF1 targets AKT to the UPS-mediated degradation, and thereby activates GSK3B. Activated GSK3B phosphorylates CRMP2 to destabilize microtubule assembly in the axons. Inhibition of this pathway results in axonal protection against degeneration
8.3 Key Molecules Involved in the Regulation of Wallerian Degeneration Process
A much clearer understanding of the cellular and molecular basis of axonal degeneration during development, and in some neurological conditions, has been elucidated in the past decade (Conforti et al. 2014; Neukomm and Freeman 2014). Next we introduce a general overview of some well-established and some newly discovered pathways that control the progression of Wallerian degeneration (Fig. 8.1b).
8.3.1 NMNAT Proteins
The interest in the mechanisms underlying Wallerian degeneration has grown dramatically since the discovery of the wlds mice (Table 8.1) (Coleman and Freeman 2010; Fang and Bonini 2012; Wang et al. 2012). The wlds mutation comprises an 85-kb tandem triplication, and subsequent cloning of the mutation has revealed the presence of a genomic triplication, generating a unique fusion protein called WldS protein, consisting of the first 70 amino acid residues of the ubiquitin-chain elongation factor E4b fused to the complete sequence of NMNAT1 and a brief linker sequence (Coleman et al. 1998; Conforti et al. 2000; Mack et al. 2001). We and others have previously shown that overexpression of NMNAT enzymatic activity, which mediates NAD synthesis, is essential for the axonal protection phenotype observed in wlds mice (Araki et al. 2004; Sasaki et al. 2009a; Babetto et al. 2010). In addition to NMNAT1, both NMNAT2 (located mainly in Golgi apparatus) and NMNAT3 (located mainly in mitochondria) have also been identified in mammals (Berger et al. 2005). Neuronal expression of the WldS protein and NMNAT3 but not NMNAT1 protein in vivo, protects axons from Wallerian degeneration, and suggests that mitochondrial expression of NMNAT activity may play a role in the mechanism of axonal protection observed in wlds mice (Yahata et al. 2009). Thus, the subcellular localization of the WldS protein is thought to be one of the important determinants for its role in axonal protection.
Expression of WldS, or other NMNATs, markedly delays axonal degeneration triggered by a variety of physical or toxic insults (Table 8.1) (Coleman and Freeman 2010; Wang et al. 2012; Conforti et al. 2014). These observations suggest that NMNAT enzymatic activity is required for axonal protection, but the correlation between neuronal NAD levels and axonal survival is lacking so far. For instance, high concentrations of exogenous NAD are reported to delay axonal degeneration in the in vitro Wallerian degeneration model (Araki et al. 2004; Wang et al. 2005; Sasaki et al. 2006), but increasing NAD levels in mice by removing the NAD consuming enzyme poly-ADP ribose polymerase 1, which catalyzes the NAD-dependent addition of ADP-ribose polymers to a variety of proteins, gives no protective effects on axonal degeneration (Sasaki et al. 2009b). Furthermore, pharmacological inhibition of nicotinamide phosphoribosyltransferase, which lowers intracellular NAD level, provides only moderate effects against axonal degeneration, and does not affect NMNAT-mediated axonal protection either (Sasaki et al. 2009b; Shen et al. 2013). Thus, the exact mechanism by which the WldS protein or NMNAT proteins suppress axonal degeneration has not been fully elucidated.
8.3.2 SARM1
Drosophila sterile α- and armadillo-motif-containing protein (dSARM) has been found through the extensive screening of ethyl methanesulfonate-treated fruit flies, to provide strong axonal protection (Osterloh et al. 2012). Deletion of the SARM1 protein in mice is sufficient to protect axons from degeneration for days after peripheral nerve transection, to an extent similar to that observed in the wlds axons (Osterloh et al. 2012). SARM1 is the fifth Toll/IL-1 receptor (TIR) domain-containing adaptor protein identified to regulate Toll-like receptor downstream signaling, and has therefore been studied with respect to innate immunity, and has been shown to control the expression of several inflammatory cytokines (Lin et al. 2014). Unlike the other TIR domain-containing adaptor proteins, SARM1 is predominantly expressed in neurons in the brain and promotes neuronal death after oxygen-glucose deprivation (Kim et al. 2007; Chen et al. 2011; Lin et al. 2014). These observations suggest that SARM1 regulates the innate immune responses of the CNS through regulating cytokine expression by neurons. By contrast, Sarm1/tir-1 has been shown to regulate the left–right asymmetric expression of odorant receptor genes in olfactory neurons in nematoda (Chuang and Bargmann 2005). In mammals, SARM1 has been also shown to play important roles in controlling neuronal morphology, via the interaction with a transmembrane heparan sulfate proteoglycan syndecan-2 to induce the mitogen activated protein kinase, kinase 4–c-Jun amino-terminal kinase (JNK), pathway. Further studies are needed to elucidate a possible mechanism(s) for the SARM1-mediated axonal protection effect.
8.3.3 Calpains and Other Proteases
Protease activation – such as in calpains – has also been strongly implicated in axonal degeneration (George et al. 1995; Ikegami et al. 2004; Gerdts et al. 2011). For example, inhibition of calpains delays axonal degeneration induced by vinblastine or by NGF withdrawal (Gerdts et al. 2011). Calpains are Ca2+-activated cysteine proteases linked to a variety of physiological processes, and their activity is critically controlled, not only by intracellular Ca2+, but also by the endogenous inhibitor calpastatin (Goll et al. 2003; Liu et al. 2004). Endogenous calpastatin has been proposed to play pro-survival roles in adult neurons under degenerative conditions, including in models of brain ischemia (Bano et al. 2005; Takano et al. 2005; Rao et al. 2008; Vosler et al. 2011; D’Orsi et al. 2012) and MPTP/6-hydroxydopamine (6OHDA)-induced PD models (Crocker et al. 2003; Grant et al. 2009). Recently, exogenous transgenic expression of calpastatin in mice provided definitive evidence of calpain involvement in sciatic and optic nerve degeneration after transection (Yang et al. 2013). Calpastatin depletion was observed in degenerating axons, while maintaining calpastatin in transected axons inhibited degeneration in vitro, and in the optic nerve in vivo. Calpastatin depletion also occurred in a caspase-dependent manner in trophic factor-deprived sensory axons in vitro, and was required for this model of developmental degeneration. These findings suggest that inhibition of calpain activity by calpastatin is one of the critical steps for the progression of axonal degeneration and calpain activation can be implicated in neurodegenerative disorders (Liu et al. 2004; Araujo and Carvalho 2005; Camins et al. 2006).
8.3.4 N-APP and DR6
Signaling components, including the Fas death receptor (CD95), tumor necrosis factor (TNF) receptors, and the p75 receptor involved in developmental axonal destruction, are known to be observed in the brain of neurodegenerative disorders (Haase et al. 2008). However, as these signaling pathways are also upregulated in response to neuronal injury, it is uncertain whether these components contribute to primary causative mechanisms or if they reflect secondary processes such as neuroinflammation. Nikolaev et al. reported a physiological function for the amyloid precursor protein (APP)-processing product N-APP in developmental axonal pruning and neuronal apoptosis, in which N-APP binds directly to a death receptor to trigger the culling of axons and neurons during development, and possibly in disease-associated neurodegeneration as well (Nikolaev et al. 2009). Using in vitro axonal degeneration and genetic mouse models, they determined that death receptor 6 (DR6) is required for these two self-destructive processes both in vivo and in trophic factor-deprived neuronal cultures, and they found that DR6-dependent axonal pruning is mediated by caspase 6, and neuronal apoptosis by caspase 3.
8.3.5 UPS and E3 Ligases
One of the first mechanistic insights into the subcellular signaling of axonal degeneration was the demonstration of the involvement of ubiquitin-proteasomal protein degradation system (UPS) in the progression of axonal degeneration. Zhai et al. showed that the application of a pharmacological inhibitor for UPS can cause delayed axonal degeneration of transected axons both in vitro and in the optic nerve in vivo (Zhai et al. 2003). However, the exact E3 ubiquitin ligase required for the progression of axonal degeneration in mammals, has long been unclear. We and others identified the E3 ubiquitin ligases and ZNRF1 and PAM-Highwire-Rpm-1 (PHR1) as the critical regulators for axonal degeneration, respectively.
8.3.5.1 ZNRF1
We previously identified ZNRF1 protein as an E3 ligase that is widely and constitutively expressed in neurons during development, as well as in adulthood (Araki and Milbrandt 2003). Interestingly, overexpression of a dominant-negative form of ZNRF1 can prevent Wallerian degeneration. These observations raise the possibility that ZNRF1 is an E3 ligase involved in the regulation of Wallerian degeneration. During the functional analysis of the UPS in axons during degeneration, we noticed that the expression level of protein kinase B (AKT) and its enzymatic activity, showed significant decreases during degeneration, and that UPS is involved in the downregulation of AKT. From these findings, we hypothesize that ZNRF1-mediated protein degradation is involved in the post-translational regulation of AKT expression. We therefore examined the possibility that ZNRF1 mediates AKT degradation in degenerating axons, and found that ZNRF1 promotes Wallerian degeneration by causing AKT to degrade via the UPS (Fig. 8.1c) (Wakatsuki et al. 2011). AKT phosphorylates glycogen synthase kinase 3B (GSK3B) and thereby inactivates it in axons. AKT overexpression significantly delays axonal degeneration. Overexpression of the active form of GSK3B induces collapsin response mediator protein 2 (CRMP2) phosphorylation at 514th Threonine (T514), which is required for the microtubule reorganization observed in the degenerating axon. The inhibition of GSK3B and the overexpression of non-phosphorylated CRMP2, both protected axons from Wallerian degeneration. These observations indicated the molecular identity of the ZNRF1-mediated regulation of kinase signaling that controls cytoskeletal integrity during Wallerian degeneration.
8.3.5.2 PHR1
Deoxyribodipyrimidine photo-lyase (PHR1) is an evolutionarily conserved family of large E3 ubiquitin ligases that are central regulators of axonal biology. Mutations of PHR1 orthologs from worms to mice lead to dramatic defects in the development of synapses and axons (Schaefer et al. 2000; Bloom et al. 2007; Lewcock et al. 2007). In invertebrates, PHR1 proteins accelerate the regenerative response following nerve injury, and delay synapse loss in genetic models of cytoskeletal instability (Xiong et al. 2010; Nix et al. 2011). In mice, loss of PHR1 results in the prolonged survival of severed axons, both in the PNS and CNS, as well as the preservation of motor and sensory nerve terminals (Babetto et al. 2013). Interestingly, PHR1 promotes self-destruction through control of the expression levels of NMNAT2 in injured axons (Xiong et al. 2012). These observations suggest that pharmacological inhibition of PHR1 function may be an attractive therapeutic candidate for ameliorating axonal loss in neurodegenerative diseases.
8.4 ZNRF1 Activation Induces Neuronal Apoptosis
As described previously, CRMP2 degradation, induced by its phosphorylation at T514 (CRMP2 pT514), leads to microtubule destabilization, and thereby promotes axonal degeneration. Therefore, CRMP2 pT514 can be an indicator for the activation of ZNRF1-mediated signaling in neurons. CRMP2 pT514 is often observed in neurons in animal models, and in patients with brain ischemia or neurodegenerative diseases (Ryan and Pimplikar 2005; Cole et al. 2007; Hou et al. 2009; Williamson et al. 2011). To show that ZNRF1-mediated signaling is activated in neurons under oxidative stress, we chose to examine a focal cerebral ischemia model. Focal ischemia is known to cause different types of cell death; neurons in the ischemic core undergo necrotic cell death, whereas neurons in the ischemic penumbra surrounding the ischemic core, mostly show delayed neuronal apoptosis, and oxidative stress is strongly implicated in the latter (Ueda and Fujita 2004; Broughton et al. 2009). Using a middle coronary artery occlusion (MCAo) model in adult male mice, we found increased CRMP2 pT514 immunoreactivity in neurons in the ischemic penumbra but not in the infarct core (data not shown). These results raised the possibility that ZNRF1-mediated AKT degradation is involved in the regulation of oxidative stress induced neuronal apoptosis. To show that the ZNRF1–AKT–GSK3B–CRMP2 pathway can be involved in the regulation of oxidative stress-induced pathology in the nervous system, we examined the significance of ZNRF1–AKT–GSK3B–CRMP2 pathway activation in oxidative stress-induced pathology, and found that the up-regulation of ZNRF1 activity by oxidative stress is sufficient to trigger neuronal degeneration and the prevention of phosphorylation-induced ZNRF1 activation protects neurons from both axonal degeneration and apoptosis (Wakatsuki et al., unpublished results). Next, we discuss our recent findings, which show that ZNRF1 activation induces neuronal apoptosis.
8.4.1 ZNRF1 Is Activated by Oxidative Stress in Neurons
Application of 6OHDA has frequently been used as an inducer of oxidative stress in cultured neurons, as well as in animal tissues (Grunblatt et al. 2000; Blandini and Armentero 2012). To examine whether ZNRF1 is involved in oxidative stress-induced neuronal apoptosis, we assessed the expression levels of AKT in 6OHDA-treated primary cultured cortical neurons overexpressing wild-type ZNRF1, a dominant-negative form of ZNRF1 C184A, or under RNAi-mediated ZNRF1 down-regulation (Fig. 8.2). We found that AKT degradation is induced in 6OHDA-treated neurons. Importantly, AKT degradation was prevented by the overexpression of ZNRF1 C184A to an extent similar to the treatment of the proteasome inhibitor MG132, but was not affected by the overexpression of wild-type ZNRF1 (Fig. 8.2a, b). In neurons expressing shRNA for ZNRF1, we also observed that AKT expression levels are (relatively) maintained compared to the control (data not shown). These results suggest that ubiquitin ligase activity of ZNRF1 is induced by oxidative stress in neurons.
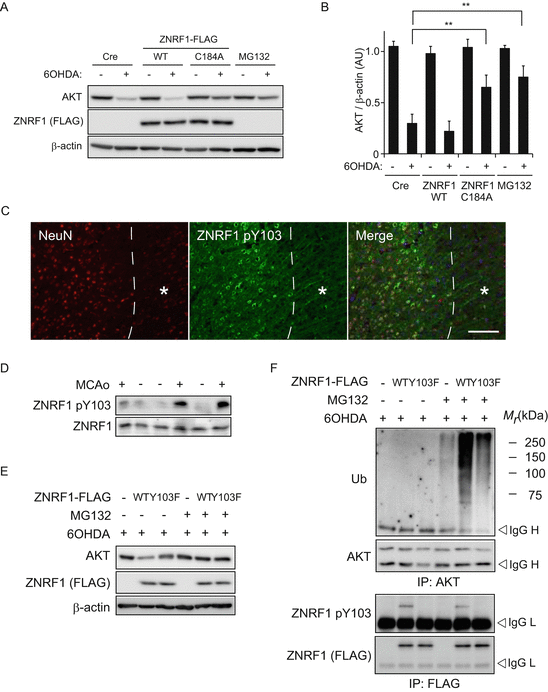
Fig. 8.2
Phosphorylation of ZNRF1 at Y103 in response to oxidative stress correlates with the up-regulation of its ubiquitin ligase activity toward AKT protein. (a, b) ZNRF1 expression promoted UPS-mediated AKT degradation in 6OHDA-treated neurons. Cultured cortical neurons were infected with adenovirus vector expressing myc-tagged wild-type ZNRF1 or dominant-negative ZNRF1 C184A mutant for 48 h and maintained with or without 20 μM 6OHDA for 3 h to induce neurotoxicity. After culture for 16 h in the presence or absence of proteasome inhibitor MG132, cell lysates were prepared from each culture and analysed by immunoblot analysis using antibodies against AKT or myc-tag (for ZNRF1). Cre only-expressing neurons or MG132-treated neurons served as negative and positive controls, respectively. β-actin served as the loading control. Representative immunoblot (a) and quantified expression levels for AKT normalized to β-actin are shown in b in comparison with control culture (Cre, +6OHDA) (mean ± SEM, five independent experiments). The asterisks indicate significant difference (P < 0.001) from control. (c) Phosphorylation of ZNRF1 at Y103 in neurons in the MCAo animal model. Representative photomicrographs for immunostaining of a neuron-specific marker NeuN or ZNRF1 pY103 on coronal sections after 4 h of MCAo. The dashed line indicates a border between ischemic penumbra and infarct core. Scale bar = 50 μm. Note that ZNRF1 pY103 immunoreactivity is observed in the penumbral neurons in sharp contrast with the absence of its immunoreactivity in the infarct core (asterisk). (d) Cell lysates were prepared from tissue sections of the ipsilateral (+) or the contralateral (−) striatum in MCAo animal model after 4 h of occlusion, and subjected to immunoblot analysis using antibodies against ZNRF1 pY103 or ZNRF1. (e, f) Wild-type (WT) or phosphorylation-resistant form of ZNRF1 Y103F mutant (Y103F) expressing cultured cortical neurons were treated with 20 μM 6OHDA for 3 h and maintained in the presence or absence of MG132 for 16 h. Cre only-expressing neurons served as a negative control (labeled as “-”). (e) Protein expression in 5 % input of cell lysates was confirmed by immunoblot analysis using antibodies against AKT or FLAG-tag (for ZNRF1) in a bottom panel. β-actin served as the loading control. (f) Cell lysates were subject to immunoprecipitation using antibodies against AKT, and the resultant immunoprecipitates were analyzed by immunoblot using antibodies against ubiquitin or AKT, respectively (top). With regard to ZNRF1-FLAG WT or Y103F, the immunoprecipitates using anti-FLAG antibody were analyzed by immunoblot using antibodies against FLAG-tag (for ZNRF1) or ZNRF1 pY103 (middle)
8.4.2 ZNRF1 Is Activated by Its Phosphorylation at Y103
As described previously, the ZNRF1 protein is widely and constitutively expressed in neurons during development as well as in adulthood (Araki and Milbrandt 2003). This suggests that, whereas we previously showed that ZNRF1–AKT–GSK3B–CRMP2 is a major signaling pathway in the promotion of Wallerian degeneration, ZNRF1 expression is not sufficient to induce Wallerian degeneration (Wakatsuki et al. 2011). It has been reported that some E3 ligases are not constitutively active, but are subject to regulation by their post-translational modifications including phosphorylation (Zhong et al. 2005; Gallagher et al. 2006). We therefore examined the possibility that ZNRF1 activity is regulated by its phosphorylation. Interestingly, ZNRF1 was phosphorylated at tyrosine residue(s) in SHSY5Y neuroblastoma treated with 6OHDA. Employing a web-based program that predicts potential tyrosine phosphorylation site(s) of ZNRF1 and the cognate kinases (NetPhos and NetPhosK, respectively), we found that Y103 can be phosphorylated by receptor tyrosine kinases including epidermal growth factor receptor tyrosine kinase (EGFR). Using antiserum against phosphorylated ZNRF1 at Y103 (ZNRF1 pY103), we found that ZNRF1 pY103 is induced by 6OHDA treatment in cultured cortical neurons in both dose- and time- dependent fashion (data not shown) and is also observed in in vivo settings, including a mouse model of 6OHDA-induced brain lesion (data not shown) and the MCAo model (Fig. 8.2d). To show that ZNRF1 pY103 is linked to its ubiquitin ligase activity, we examined AKT ubiquitination in 6OHDA-treated cultured cortical neurons overexpressing wild-type ZNRF1 or ZNRF1 Y103F mutant. We found that the expression level of AKT was significantly decreased in neurons expressing wild-type ZNRF1, but not in the ZNRF1 Y103F mutant (Fig. 8.2e, f). AKT ubiquitination, on the other hand, was increased in wild-type ZNRF1-expressing cells, but not in cells expressing the ZNRF1 Y103F mutant. These results suggest that ZNRF1 pY103 results in activation of the ubiquitin ligase activity. Importantly, EGFR inhibition by compound 56 – a potent inhibitor of EGFR – significantly inhibited ZNRF1 phosphorylation and AKT ubiquitination (data not shown). These results indicate that ZNRF1 is specifically phosphorylated at Y103 in neurons in response to oxidative stress, and that the oxidative stress-induced ZNRF1 pY103 is mediated by EGFR.
8.4.3 Inhibition of ZNRF1 Activation Protects Primary Cultured Neurons from Oxidative Stress-Induced Apoptosis
To examine whether activation of ZNRF1 by phosphorylation at Y103 in response to oxidative stress in neurons leads to apoptosis, we assessed the expression of cleaved caspase 3, a marker for prototypical apoptosis pathway activation, in 6OHDA-treated cultured cortical neurons overexpressing ZNRF1C184A or ZNRF1 Y103F. We found that 6OHDA-induced activation of the apoptosis signaling pathway was blocked by expression of these mutant forms of ZNRF1, suggesting that ZNRF1 activation in neurons by oxidative stress resulted in increased apoptosis signaling. To further analyze subcellular signaling downstream of ZNRF1, we examined the expression of cleaved caspase 3 in 6OHDA-treated cultured cortical neurons overexpressing myristoylated form of AKT (myrAKT; a constitutively active form of AKT) or kinase-dead form of GSK3B (GSK3B K85M) (Fig. 8.3a, b). We found that expression of myrAKT and GSK3B K85M both prevented 6OHDA-induced apoptosis. These results suggested that AKT–GSK3B kinase cascade is involved in ZNRF1 activation-induced neuronal apoptosis signaling. Similar results were obtained in multiple different types of neurons, such as SHSY5Y neuroblastoma or the ventral mesencephalic neurons cultures, which is known to be rich in dopaminergic neurons (data not shown). These results suggest that oxidative-stress induced activation of ZNRF1 in neurons, results in apoptosis via ZNRF1-dependent proteasomal degradation of AKT, and resultant activation of GSK3B.
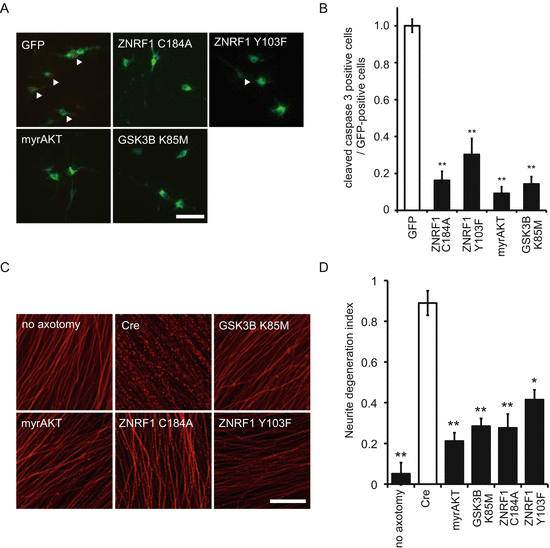
Fig. 8.3
 Parkinson’s Disease; Neurodegeneration as Systemic Disease
Parkinson’s Disease; Neurodegeneration as Systemic Disease
 Brain–Peripheral Organ Communication
Intermediate Phenotype Approach for Neuropsychiatric Disorders
Social Implementation of Neurodegenerative Disease Research and Neuroethics
Brain–Peripheral Organ Communication
Intermediate Phenotype Approach for Neuropsychiatric Disorders
Social Implementation of Neurodegenerative Disease Research and Neuroethics
 Clinical Systems Neuroscience
Clinical Systems Neuroscience
 Home- and Community-based Medical Care for Neurodegenerative Diseases: ALS as an Illustration
Home- and Community-based Medical Care for Neurodegenerative Diseases: ALS as an Illustration




Phosphorylation of ZNRF1 at Y103 is involved in the progression of Wallerian degeneration and oxidative stress-induced neuronal apoptosis. (a, b) Oxidative stress-induced neuronal apoptosis is not observed when ZNRF1 phosphorylation at Y103 is inhibited in cultured primary cortical neurons. Cleaved caspase 3 immunofluorescence was assessed to determine the effect of AKT, GSK3B, ZNRF1 or their mutant expression. Representative photomicrographs for cleaved caspase 3 immunostaining are shown in (a). Open arrowheads indicate cleaved caspase 3-positive cells. (b) The ratio of the cleaved caspase 3-positive cell number to total number of GFP-positive cells for each condition are shown (mean ± SEM, five independent experiments). The asterisks indicate significant difference (P < 0.001) from control (open bar, labeled as “GFP”). (c, d) Neurite protection effect by ZNRF1–AKT–GSK3B pathway molecules was assessed by in vitro Wallerian degeneration model using neurites from cultured DRG explant neurons overexpressing indicated proteins via adenoviral vector infection. Representative photomicrographs of degenerating neurites from neurons expressing indicated genes using adenoviral vectors at 24 h after initiation of Wallerian degeneration are shown in c. Scale bar = 25 μm. Neurite degeneration index values calculated for each condition at 24 h are shown in d (five independent experiments). The asterisks indicate significant difference (*P < 0.05 and **P < 0.01) from Cre-only expression control (labeled as “Cre”)
Related posts:
Parkinson’s Disease; Neurodegeneration as Systemic Disease
Brain–Peripheral Organ Communication
Intermediate Phenotype Approach for Neuropsychiatric Disorders
Social Implementation of Neurodegenerative Disease Research and Neuroethics
Clinical Systems Neuroscience
Home- and Community-based Medical Care for Neurodegenerative Diseases: ALS as an Illustration
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
