Fig. 11.1
Relative risk of subsequent glioma (closed boxes) and meningioma (open boxes) within the Childhood Cancer Survivor Study cohort by radiation dose (Printed with permission from Oxford University Press, (Neglia et al. 2006))
In a cohort of ALL survivors followed by St Jude Children’s Research Hospital, ten patients of the 1,612 survivors developed subsequent high-grade gliomas (glioblastoma multiforme, n = 4, anaplastic astrocytoma, n = 2, other high grade glioma, n = 4) and one patient developed a low-grade glioma. Of those developing high grade tumors, latency from the time to diagnosis to tumor development ranged from 5.9 to 14.1 years (median 9 years) (Walter et al. 1998). Similar to the NA-CCSS cohort, children less than 6 years at the time of initial cancer diagnosis were more likely to develop high-grade tumors, as were children with CNS disease at the time of diagnosis.
Secondary Meningioma
Meningiomas are typically thought of as benign tumors arising from the dura mater surrounding the brain and spinal cord (see Fig. 11.2). The association with cranial radiation has been understood for quite some time, as these lesions have occurred at higher rates in children treated with radiation for tinea capitis (Ron et al. 1988; Sadetzki et al. 2002).
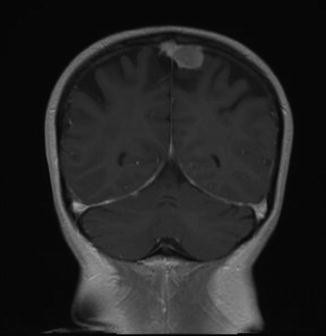
Fig. 11.2
Para-falcine meningioma in a 20+ year survivor of childhood acute lymphoblastic leukemia
In the NA-CCSS cohort, meningiomas occurred much later (median 22.9 years, range 15.8–32.7 years) than either subsequent glial tumors or medulloblastomas. This finding was similar to the St. Jude ALL cohort, where meningiomas were diagnosed a median of 19 years from initial cancer diagnosis (Walter et al. 1998), as well as the BCCSS cohort, where meningiomas were diagnosed 23.1 years from initial cancer diagnosis (Taylor et al. 2010).
Among the NA-CCSS cohort, meningioma risk was increased by female sex, younger age at primary diagnosis, radiation therapy exposure and previous treatment for CNS tumor. They occurred at the highest rate among survivors of medulloblastoma, with cumulative incidence of 16.4 % (95 % CI = 7.5–25.3 %) (Friedman et al. 2010). Multiple studies have attempted to establish associations between chemotherapy exposure and subsequent meningioma, and for the most part, no associations have been found; however, Taylor et al. (2010) reported an interesting association between intrathecal methotrexate and risk of meningioma, in an analysis of secondary CNS tumors in the BCCSS. Within the BCCSS cohort, individuals who had received at least 70 mg/m2 of intrathecal methotrexate had a 36-fold increased risk of meningioma when compared to those unexposed. Within this same cohort, no relationship was observed with systemic methotrexate or other agents. Other studies have failed to find an association between intrathecal chemotherapy and meningioma (Fontana et al. 1987; Walter et al. 1998). In a 2006 analysis of the NA-CCSS patients who had follow-up through 2002, a detailed reconstruction of radiation exposure, including location and dose, was performed to better understand the dose-response relationship between radiotherapy and the development of CNS tumors. Within this cohort, 66 meningiomas were identified, of which three were classified as malignant with other subtypes classified as: meningotheliomatous (5), fibrous (4), and transitional (4) (Neglia et al. 2006). Meningiomas were more common than gliomas in those with secondary CNS tumors. While the overall median time to secondary CNS tumor development was 14 years, it was 17 years for those patients developing meningiomas, with 71 % of patients being diagnosed 15 years or more after their initial cancer diagnosis. The majority of this subset of patients (74 %) was diagnosed after 20 years of age. In children exposed to radiation therapy for their primary malignancy, a positive linear dose-response relationship was observed for development of a meningioma. For survivors treated with 10–20 Gy, the odds of developing meningioma were 12, whereas in those treated with 20–30 Gy the odds increased to 21.6 and, most notably, in survivors who received radiation doses greater than 30 Gy, the odds of developing a meningioma was in the order of 50–100 times that of the general population (see Fig. 11.1).
In an Israeli study of 210 patients treated for acute lymphoblastic leukemia and T-cell non-Hodgkin’s lymphoma before 1990, meningiomas were detected in 16 survivors. Median age of detection was 28.7 years and median time from diagnosis was 21 years (10–29 years). Of the 210 patients, 88 had received cranial radiation, and of the 16 identified meningiomas, 15 had received cranial radiation at ages 2–14 years (median 7.6 years); 14 had received 24 Gy and one had received a 18 Gy. This group found the rate of meningiomas increased after 15 years from therapy (Goshen et al. 2007).
In a unique genetic case-control study, a cohort of patients who had been treated with cranial radiation for tinea capitis and then gone on to develop subsequent meningioma was recruited for participation. Their family members, both exposed and not exposed to radiation, were recruited as well. Patients who had been treated with radiation and had not developed subsequent meningioma were recruited, with their family members, as well. Radiation exposure was variable within this group (estimated average dose range to the brain 1.0–6.0 Gy). The patients with radiation-associated meningiomas were more likely to have one or more first-degree relatives develop meningioma as compared to the radiated patients who had not developed meningiomas. These findings suggested the possibility of genetic susceptibility to developing tumors following radiation exposure, although specific candidate genes were not identified and similar studies would be nearly impossible to carry out (Flint-Richter and Sadetzki 2007).
With longer follow-up, meningiomas are now being reported in excess of malignant brain tumors, confirming the longer latency to development (Friedman et al. 2010) and the need for long-term follow up of childhood cancer survivors. There is some evidence that the incidence of meningiomas is underestimated by as much as one-third, since they can often be asymptomatic and routine screening is not part of standard follow-up care (Larjavaara et al. 2008).
Screening
Currently the United Kingdom does not recommend routine imaging or surveillance for childhood cancer survivors previously treated with cranial radiation (CCLG 2005). The North American-based Children’s Oncology Group follow-up guidelines recommend screening brain MRIs beginning 2 years after completion of radiotherapy for survivors with neurofibromatosis (NF) and for symptomatic survivors at high risk of developing second brain tumors (COG 2008). Presently, the majority of subsequent neoplasms of the CNS are identified once they have become symptomatic or incidentally during investigation of another issue.
Survival
There are limited data available on survival following secondary neoplasms of the CNS among childhood cancer survivors. Treatment can be challenging, particularly in higher grade tumors, because of the risks associated with re-irradiation of the CNS. One of the first studies to investigate survival by CNS tumor type was conducted by the BCCSS (Taylor et al. 2009). In their cohort, 13,211 survivors were surveyed and 247 tumors of the brain and spine were identified as secondary neoplasms; of these, 73 were gliomas, 137 meningiomas, and the rest were a variety of other tumors types. Of these 247 second CNS neoplasms, 123 led to death, 62 after glioma and 42 after meningioma; 73 % of the deaths were classified as death from secondary neoplasm. Five year relative survival following meningioma was greater than 80 % for both males and females while for glioma was only 19.5 % with worsening survival paralleling higher tumor grade. Worse outcomes were also observed in patients treated in earlier treatment eras. In a smaller cohort of 1,612 ALL survivors from St. Jude Children’s Research Hospital, 22 subsequent CNS tumors were identified; 11 of which were meningiomas, 10 high-grade gliomas, and one low-grade glioma. Overall survival following meningiomas was very good, with all patients alive with up to 10 years of follow-up. Prognosis in the high-grade glioma group was very poor (Walter et al. 1998).
Related posts:
 Hyperinsulinemia Tends to Induce Growth Without Growth Hormone in Children with Brain Tumors After Neurosurgery
Hyperinsulinemia Tends to Induce Growth Without Growth Hormone in Children with Brain Tumors After Neurosurgery
 Drugs for Primary Brain Tumors: An Update
Drugs for Primary Brain Tumors: An Update
 Survivors of Childhood Cancer: Risk of New Primary Neoplasms of the CNS
Survivors of Childhood Cancer: Risk of New Primary Neoplasms of the CNS
 Treatment of Brain Tumors: Electrochemotherapy
Treatment of Brain Tumors: Electrochemotherapy
 Brain Metastases: The Application of Stereotactic Radiosurgery and Technological Advances
Brain Metastases: The Application of Stereotactic Radiosurgery and Technological Advances
 Nonthermal Irreversible Electroporation as a Focal Ablation Treatment for Brain Cancer
Nonthermal Irreversible Electroporation as a Focal Ablation Treatment for Brain Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


