Fig. 12.1
Time to recurrence of subsequent glioma (open bars) and meningioma (closed bars) in the Childhood Cancer Survivor Study cohort (Printed with permission from Oxford University Press (Neglia et al. 2006))
There is suggestion that since some tumor types, such as meningioma, can behave in a relatively benign manner, they may be underreported. Estimates have placed incidence as much as one-third higher than reported figures (Larjavaara et al. 2008). This is addressed in an Israeli study of survivors of childhood ALL or T-cell lymphoma in which the survivors were followed with serial cranial imaging (either CT or MRI), every 3–6 years. The patient population included both children who had been treated with cranial radiation (n = 76) and children who had not received radiation (n = 74). The investigators observed 16 meningiomas, with median time of 21 years from initial tumor diagnosis; fifteen of the survivors who developed meningioma had previously received radiation therapy. Of the sixteen meningiomas, only one had clinical symptoms (seizures); the remaining were diagnosed because of screening. Of patients who had received cranial radiation and remained available for screening, cumulative incidence was remarkably high (53.8 %) at 25 years from cranial radiation exposure (Goshen et al. 2007).
Therapeutic Risk Factors
Radiation therapy has been recognized as a risk factor for development of subsequent CNS tumors for quite some time; as a result of this and other considerations, the proportion of children receiving radiotherapy as part of their initial cancer therapy has decreased over time, with 56 % of children receiving it in 1973–1979, to only 28 % in 1995–2002 (Inskip and Curtis 2007). In an analysis of 446 children treated with megavoltage radiation at the University of Minnesota, 37 subsequent neoplasms were identified, including 6 meningiomas and 2 astrocytomas, all of which developed within the initial radiation field. This represented a 4 % (95 % CI = 2–8 %) cumulative risk of developing a brain tumor at 30 years of follow-up (Gold et al. 2003). Goshen and colleagues identified 16 meningiomas in their population of 150 childhood cancer survivors and all but one of those individuals had been previously treated with cranial radiation (Goshen et al. 2007). Similarly, in the SJCRH cohort of 1612 ALL survivors, 21 survivors developed subsequent CNS tumors and all had previously been treated with radiation therapy (Walter et al. 1998). Within the CCSS cohort, second neoplasms were higher among those treated with radiation therapy, with a relative risk of 2.7 (95 % CI = 2.2–3.3) (Friedman et al. 2010). Similarly, within the GCCR cohort of ALL survivors, all secondary CNS tumors except one survivor who developed meningioma had received cranial radiation at some point during therapy. The risk of developing a subsequent CNS tumor was 0.1 % for the non-radiated survivors and 1.3 % for the radiated group and an increased risk was noted with increased radiation dose (Loning et al. 2000).
Highly significant radiation dose–response relationships have been observed in multiple cohorts (see Fig. 12.2). Within the NA-CCSS cohort, this relationship was observed when all CNS tumors were combined and persisted when individual tumor histologies were examined separately; specifically, the odds of developing glioma rose across radiation categories to a peak of 21 for the 30–44.9 Gy category and the odds of meningioma peaked at 96.3 in the 30–44.9 category (Neglia et al. 2006). The risk of both meningiomas and gliomas was strongly associated with dose of radiation therapy, with an excess relative risk (ERR) of 1.06 per Gy for meningioma and 0.33 per Gy for glioma (Neglia et al. 2006). Similar linear relationships were described in the BCCSS cohort, although with different effects, both for the development of subsequent gliomas/PNETs (ERR = 0.079 per Gy) and for meningiomas (ERR = 5.1 per Gy). At a cumulative radiation dose of ≥ 40 Gy, the relative risk of developing a subsequent meningioma was 479 times that of non-radiated survivor, whereas the relative risk of subsequent glioma/PNET was 4.4 (Taylor et al. 2010). This is the highest relative risk currently reported for meningioma.
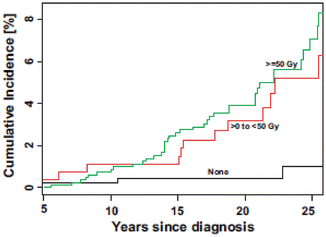
Fig. 12.2
Cumulative incidence of CNS neoplasms by cranial radiation therapy dose (Printed with permission from Oxford University Press (Armstrong et al. 2009b))
As the use of radiation therapy has decreased over time, the trend has moved in the opposite direction for chemotherapy (Inskip and Curtis 2007). With the advent of improved risk-stratification, high-risk patients are receiving more aggressive chemotherapy regimens (Hudson et al. 2012) and more aggressive intrathecal chemotherapy regimens are being used in place of prophylactic radiation in the case of ALL (Pui et al. 1998). There has been some debate regarding the association of systemic chemotherapy and the development of subsequent neoplasms in the CNS. Multiple cohort studies have looked at this issue and have reported variable results.
Within the NA-CCSS cohort, when adjusting for radiation exposure and original diagnosis, chemotherapy exposure did not appear to significantly increase the risk of second CNS tumor development, even when looking at different chemotherapy classes separately (Neglia et al. 2006; Friedman et al. 2010). Similarly, within the GCCR cohort of ALL survivors and the French-British cohort of non-leukemia childhood cancer survivors, no associations were observed between types or doses of systemic chemotherapy and development of a secondary CNS tumor (Loning et al. 2000; Little et al. 1998). Although no associations were observed between any types of systemic chemotherapy, including methotrexate, and the development of either glioma/PNET or meningioma within the BCCSS, an association between intrathecal methotrexate and subsequent CNS neoplasms has been reported. When adjusting for radiation exposure, a linear relationship was observed with the risk among those exposed to ≥ 70 mg/m2 was 36 times greater than among those who were not treated with intrathecal methotrexate (Taylor et al. 2010). Associations between intrathecal chemotherapy and risk of subsequent CNS neoplasms have previously been evaluated in the ALL population; however, no other significant associations have been identified to date (Fontana et al. 1987; Walter et al. 1998).
One of the few studies to identify a link between systemic chemotherapy and subsequent CNS tumors was published by Relling et al. (1999) from SJCRH. They identified an increased rate of secondary malignant brain tumors in children receiving therapy on one of their research protocols which gave more intensive antimetabolite therapy before and during radiation therapy. Of the 153 children enrolled, 52 had received cranial radiation, and of those, 6 (12.8 %) developed brain tumors within 7–10 years from the time of radiation. Tumor histologies included: glioblastoma multiforme (3), anaplastic astrocytoma (2) and PNET (1). None of the non-radiated children developed a secondary CNS tumor. Additionally, they determined that 4/6 patients had elevated red blood cell concentrations of thioguanine nucleotide compared to the rest of the irradiated group; furthermore, among the 7 children with a genetic defect in thiopurine methyltransferase (TPMT) activity, cumulative risk of brain tumor development was 42.9 % as compared to 8.3 % in the 45 children with wild-type activity. No other therapy-related associations were identified. These findings raised concern about the possibility of antimetabolites contributing to tumor development when given concurrently with radiation; however, these findings have not been confirmed to date.
The treatment era in which a child was initially diagnosed and treated for their primary cancer has also been associated with risk of second CNS tumors in some cohorts. In the NA-CCSS cohort, survivors treated in 1975–1979 and 1980–1986 had decreased risk of second cancers as compared to those treated in 1970–1974, suggesting that changes are being seen with more conservative radiation, particularly in the ALL treatment protocols, but also recognizing that shorter follow up may be partially responsible for this finding (Friedman et al. 2010). Interestingly, when looking specifically at gliomas, higher SIR was observed for survivors treated in the most recent period of subject accrual (for diagnosis between 1980 and 1986, SIR = 12.7) (Neglia et al. 2006). Data is not available for patients being treated on contemporary treatment protocols which include more exposure to anthracyclines and cyclophosphamide. Long-term follow up is needed to determine whether risk or type of second cancers has significantly changed.
Host Risk Factors
Available cohort data suggest a role of age in modifying risk for subsequent CNS tumors, although findings have not been completely consistent. Bhatia et al. (2002) found that age at acute leukemia diagnosis did not impact risk of developing subsequent CNS neoplasm; however, Friedman et al. (2010) found that generally, without regard to primary cancer diagnosis, patients who were treated at a younger age (<10 years) had increased risk of developing subsequent CNS tumors. Within the SJCRH cohort of ALL survivors, diagnosis and treatment at less than 6 years of age was not associated with increased risk of secondary brain tumors overall, but was associated with an increased risk of developing a high grade glioma (Walter et al. 1998). Similarly, in the Children’s Cancer Study Group, children who were 5 years or younger at the time of diagnosis had a significantly higher risk of developing a secondary brain tumor as compared to those older than 5 years at diagnosis (Neglia et al. 1991).
Loning and colleagues observed that tumors of the CNS were the most common second neoplasm among children who were less than 7 years of age at the time of initial leukemia diagnosis. The cumulative probability of developing a CNS tumor was 1.5 % (95 % CI = 0.2–2.7 %) for this subgroup as compared to risk of 0.1 % (95 % CI = 0–0.3 %) in survivors who were greater than 7 years of age at the time of diagnosis (Loning et al. 2000). Neglia et al. (2006) reported that in the NA-CCSS cohort, when comparing to the general population, standardized incidence ratios for developing glioma was highest among children diagnosed with cancer at less than 5 years of age (SIR = 14.5), and the excess relative risk per Gy was highest for individuals exposed to radiation therapy prior to age 5 years of age (ERR per Gy = 0.64) as compared to those exposed at 5–9 years (ERR per Gy = 0.1) or 10–20 years (ERR per Gy = 0.15). Within the BCCSS cohort, there was a statistically significant decline in the excess relative risk of subsequent glioma/PNET with increasing age of first exposure to radiation. They did not report an effect of age of exposure on developing a subsequent meningioma (Taylor et al. 2010).
Genetic cancer predisposition syndromes, such as Neurofibromatosis 1 and 2, as well as Gorlin’s syndrome, tuberous sclerosis and Von Hippel-Lindau syndrome are all associated with increased risk of CNS tumor development (Little et al. 1998). Other host characteristics, such as gender, have not been significantly associated with risk of CNS tumor development.
Role of Primary Cancer Diagnosis
Acute leukemias account for the highest percent of new childhood cancer diagnoses. Acute lymphoblastic leukemia, specifically, is the most common, causing 3–4 cases per 100,000 children annually (Gurney et al. 1995). Five-year survival rates exceed 85 % (Jemal et al. 2010), meaning that there are approximately 2,000 long-term survivors of childhood ALL each year (Bhatia et al. 2002). In ALL survivors treated with cranial irradiation, central nervous system tumors are among the most commonly observed second neoplasms (Neglia et al. 1991; Bhatia et al. 2002; Pui et al. 2003). Within the SJCRH cohort of ALL survivors, CNS leukemia at the time of diagnosis was associated with an increased risk of a subsequent brain tumor and also increased the risk of developing a high-grade tumor; these relationships held, even when radiation dose was controlled for within the analysis (Walter et al. 1998). Cumulative incidence of secondary brain tumors after ALL therapy has been fairly consistent across different cohort studies: 0.47 % at 10 years (Bhatia et al. 2002), 1 % at 15 years (Loning et al. 2000), 1.39 % at 20 years (Walter et al. 1998). These estimates represent an approximate tenfold increased risk of developing a brain tumor as compared to the general population (Bhatia et al. 2002). A variety of CNS tumor histologies have been observed. Bhatia et al. (2002) looked at a cohort of 8,831 children treated for ALL between 1983 and 1995 on Children’s Cancer Group (CCG) research protocols. Within the cohort, 63 s malignancies were identified, 19 of which were CNS tumors. Histologies included: glioblastomas multiforme (9), anaplastic astrocytoma (4), primitive neuroectomdermal tumors (PNET) of the brain (3), meningioma (2), and medulloblastoma (1). A study of 856 ALL survivors, treated at SJCRH between 1962–1992, compared survivors treated with radiation (n = 597) and those without (n = 259) (Pui et al. 2003). Of the 44 s neoplasms, 15 occurred in the central nervous system; 10 were meningiomas and 5 were malignant tumors of various types. Among the CCSS cohort, Neglia et al. (2006) found that childhood leukemia survivors were more likely to develop subsequent gliomas as opposed to meningiomas; however, among the SJCRH cohort, primary diagnosis of leukemia was associated with risk of meningioma (Hijaya et al. 2007).
Related posts:
 Hyperinsulinemia Tends to Induce Growth Without Growth Hormone in Children with Brain Tumors After Neurosurgery
Human Brain Tumor Growth: Role of Aquaporins
Classification/Diagnosis of Brain Tumors Using Discriminant Function Analysis
Incidence of Recraniotomy for Postoperative Infections After Surgery for Intracranial Tumors
The Role of Glutathione and the Glutathione-Linked Enzyme Systems in Brain Tumor Drug Resistance
Image-Guided Stereotactic Radiosurgery for Spinal Pathology
Hyperinsulinemia Tends to Induce Growth Without Growth Hormone in Children with Brain Tumors After Neurosurgery
Human Brain Tumor Growth: Role of Aquaporins
Classification/Diagnosis of Brain Tumors Using Discriminant Function Analysis
Incidence of Recraniotomy for Postoperative Infections After Surgery for Intracranial Tumors
The Role of Glutathione and the Glutathione-Linked Enzyme Systems in Brain Tumor Drug Resistance
Image-Guided Stereotactic Radiosurgery for Spinal Pathology
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


