Fig. 1
A wet laboratory workflow for combining non-barcoded sample pooling and targeted re-sequencing
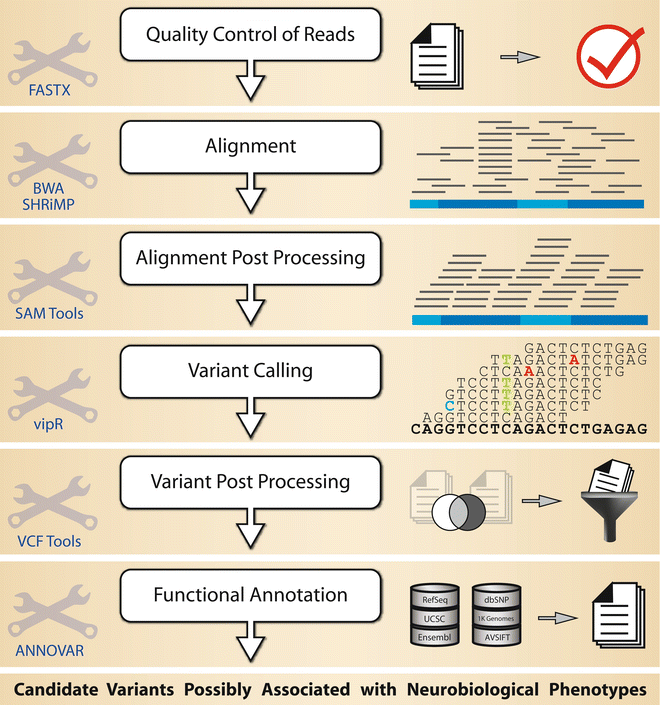
Fig. 2
Overview of the bioinformatics MPS data analysis pipeline in targeted re-sequencing with pooled samples
3.1 Non-barcoded Sample Pooling
The costs and workload for DNA sample preparation are major factors in targeted re-sequencing studies involving many individual samples. In most cases, individual sample preparation will accumulate higher costs than the actual sequencing. Thus, for reducing both labor and costs, pooling of DNAs from multiple individual biological samples can be performed prior to library preparation, that is, pooling of DNA without barcodes into a single library.
For pool sizes that reach into hundreds of people, we propose to follow a sequential pooling regime in order to maintain reasonable representations of allele frequencies throughout PCR-based enrichment. Therefore, we recommend limiting the pool sizes initially to less than 50 individuals. If there are more samples, multiple enrichment reactions may be performed in parallel that are finally combined post enrichment.
The first step in the pooling procedure is the quantification of the individual samples that will constitute the pooled library. Accurate quantification is prerequisite for a successful pooling experiment. The accuracy in absolute terms is not quite as important as the relative accuracy between the samples, i.e., values have to be comparable between the samples. Quantification of DNA can be achieved using a number of methods. We propose to quantify via fluorometry using the available dsDNA kits on the Qubit 1.0 fluorometer (Invitrogen). This approach provides an easy, quick, and accurate quantification of DNA. After quantification, the samples are combined in an equimolar fashion to constitute the pool. Naturally, the sample with the lowest amount of material will dictate the final yield of the pool. The pools are then enriched via PCR, and depending on the design there might be additional pooling steps involved post enrichment. If multiple long-range PCRs are generated from one sample pool, the obtained individual amplicons should be combined (described in detail in Sect. 3.2 and respective Notes 4.1.3 and 4.2.5). Those amplicon pooling steps can be performed as before with quantification and equimolar pooling. In some cases the PCR products can contain weak extra bands. If such artifacts occur at modest levels, one might still use the material successfully, but one needs to follow a special approach (see Notes 4.2.5).
3.2 Target Enrichment
The aim of target enrichment is to “isolate” the genomic target region. This is typically achieved by enriching the DNA in the target region so that DNA fragments of nontarget regions are represented only in negligible amounts. The definition of a target sequence is of great importance and can be determined from prior knowledge, e.g., from GWASs or in an unbiased manner focused on the most informative portion of the genome, which is believed to be the exome. Thus, after a target region is defined, e.g., all exons of a specific gene, the best-suited method depends on the size of the target region and the desired sample size (see Table 1 in Altmann et al. (2013) for a brief overview). In our protocol we used the straightforward approach using polymerase chain reaction (PCR). Briefly, primers are designed that flank the genomic loci of interest; then the regions between the primers are amplified using multiple PCRs and/or long-range PCRs. Using this method, target sizes up to 100 kb are achievable.
As generic starting point for a PCR mix, one might use a reaction volume of 25 μl with 100 ng genomic DNA, 0.8 μM of each primer, 300 μM of each deoxynucleotide, and 2.5 units of LongAmp Taq DNA polymerase. Depending on the melting temperature of the primer pair and the amplicon length, the cycling protocol should be adapted from 94 °C for 3 min as initial denaturation and then 32 cycles using 94 °C for 30 s, 59–61 °C for 40 s, and 65 °C for 2.5–11 min for 32 cycles. The final extension is carried out at 65 °C for 10 min.
For primer design sequences that harbor known variants should be avoided and the design of overlapping PCR products might be beneficial in some cases. The amplicons should be roughly equal in size and within the range of 1–15 kb. Primer design and PCR cycling conditions are standard methods, and a lot of guidelines on those topics can be found elsewhere (Abd-Elsalam 2003; Borah 2011; Davies and Gray 2002; Hogrefe and Borns 2011). In any case, a DNA polymerase with proofreading activity has to be utilized for avoiding artificial sequence mutations introduced by the PCR. Efficiency and specificity of PCR need to be evaluated by checking all amplicons on a high-resolution 0.8 % agarose gel. After PCR multiple amplicons from different loci are combined at equimolar levels in order to obtain even coverage over the whole target region. Thereby, the same procedure as for sample pooling applies, but one needs to take into consideration different lengths of amplicons in order to calculate quantities based on numbers of molecules. For targeted pool sizes greater than 50, an additional pooling of PCR pools into meta-pools is required. In this specific case quantification can be omitted since DNA amounts can be inferred from the previous measurement. Further, we recommend performing a preparative 0.8 % agarose gel from the pooled amplicons for reaction cleanup, removal of primer dimers, and removal of spurious amounts of nonspecific products. The desired PCR products are then excised from the gel, and the contained DNA is purified with standard gel purification kits.
The steps for target enrichment via PCR are the following:
1.
Identification and definition of target regions
3.3 Sequencing Using MPS
An experimental difference to individual DNA sequencing is that pools have to be sequenced with higher coverage to address the increased sample complexity of the pool. In DNA pools, the desired individual coverage is multiplied with the number of alleles in the pool. With the required total coverage and the cumulative target length, the most suitable MPS system can be selected among available MPS systems (for an overview, see Glenn (2011); please note: there are regularly updated data tables available online as mentioned in the manuscript). For smaller target sizes often the capacity of the benchtop-sized small-scale sequencers will be sufficient. In general for such projects, the large-scale systems are likely to produce more data than necessary even if only on one lane of a flow cell is used. In such a case barcoded pooling with other projects is an option. But for extremely large target regions or exome sequencing, the large-scale systems are required since otherwise the high number of sequencing runs is impracticable.
Another consideration when choosing the MPS platform for the experiment is the actual sequencing accuracy. A low raw read error rate is particularly relevant for pooled approaches since the possibility to gain accuracy by constructing consensus sequences (as done in individual sequencing) is limited. Thus, the error rate of the MPS system can directly influence the minimum allele frequency in the pool at which variants will be detectable. Unfortunately, in practice the comparison of error rates is problematic. For a discussion on the topic and a possible comparison table, see Glenn (2011).
For the library preparation a starting amount around 1 μg is advisable since this allows PCR-free library preparation. Whenever possible PCR-free library preparations should be preferred to avoid bias and allelic dropout (see Note 4.3.2). This is often feasible since PCR enrichment gives usually large yields of amplicons and additional pooling leads to accumulating total quantities.
For fragmentation of enriched long-range PCR amplicons during library preparation we observed good performance using the standard program for genomic DNA on a Covaris S2 using Covaris microTUBEs and 6 cycles of frequency sweeping mode with 20 % duty cycle, intensity 5, 200 cycles/burst, and 60 s. time. Thereby, the individual pooled amplicons should have similar length of more than 1,000 nt. Shorter amplicons of 100–600 bp might be sequenced directly without fragmentation depending on the selected MPS system. If additionally less than 20 amplicons are analyzed, then adaptations might be necessary for sequencing of such low-diversity libraries (see Note 4.3.3).
To operate the instruments for sequencing in most cases, one can adhere to the standard procedures specified by the manufacturer. Only in very specific cases one needs to implement adaptations (see Note 4.3.3). Primary analysis is usually performed on the sequencing system, and generated output files (fastq or related file formats) are finally exported for further bioinformatics analyses.
3.4 Bioinformatics Analysis
The sequences for each pool are processed separately until the variant calling step. There are substantial differences between the processing steps for standard single nucleotide variant (SNV) calling and SNV calling from pooled sequencing data. The most striking difference is that the algorithms for calling SNVs in the individual DNA from one subject can safely assume allele frequencies of either 1.0 (representing a homozygous allele) or 0.5 (representing a heterozygous allele). In contrast, allele frequencies in DNA pools may vary from 1.0 (all subjects in the pool carry the same allele) to as little as a single heterozygous allele (1/2N, with N being the number of subjects in the pool). Clearly, detecting a single heterozygous allele is a challenging task in the presence of sequencing errors. To tackle this problem we have developed the algorithm vipR (Altmann et al. 2011) for effectively discovering SNVs in pooled sequencing data. See Nielsen et al. (2011) for a review on standard SNV calling methods in DNA from individual subjects.
The processing steps are as follows:
2.
Alignment of the high-quality reads to the reference sequence using one or more short read aligners (see Note 4.4.2).
3.
Post-processing the generated alignments (see Note 4.4.3): sorting the alignment with respect to the genomic coordinates using SAMtools or Picard.
5.
Post-processing the called SNVs using VCFtools:
(a)
Remove low-confidence SNVs.
(b)
Merge SNVs called from two different alignments.
4 Notes
4.1 Non-barcoded Sample Pooling
Note 4.1.1: Limit pool size prior to PCR-based enrichment. If pool sizes become too large prior to PCR amplifications, variants with very low abundance in the pool might start to show stochastic behavior during the first PCR cycles. Hence, they destroy the true stoichiometric relations in the pool. Recently published bioinformatics approaches characterize and correct some of the biases originating from non-barcoded pooling (Chen et al. 2012).
Related posts:
 RNA Sequencing from Laser Capture Microdissected Brain Tissue to Study Normal Aging and Alzheimer’s Disease
RNA Sequencing from Laser Capture Microdissected Brain Tissue to Study Normal Aging and Alzheimer’s Disease
 An Overview of Methods Used in Neurogenomics and Their Applications
An Overview of Methods Used in Neurogenomics and Their Applications
 Location Analysis and Expression Profiling Using Next-Generation Sequencing for Research in Neurodegenerative Diseases
Location Analysis and Expression Profiling Using Next-Generation Sequencing for Research in Neurodegenerative Diseases
 Gene Expression-Based Approaches to Understanding Huntington’s Disease and New Tools for the Interpretation of Expression Datasets
Gene Expression-Based Approaches to Understanding Huntington’s Disease and New Tools for the Interpretation of Expression Datasets
 Role of Neurogenomics in the Development of Personalized Neurology
Role of Neurogenomics in the Development of Personalized Neurology
 1: Narcolepsy—Unequivocal Diagnosis After Split-Screen, Video-Polysomnographic Analysis of a Prolonged Cataplectic Attack
1: Narcolepsy—Unequivocal Diagnosis After Split-Screen, Video-Polysomnographic Analysis of a Prolonged Cataplectic Attack
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


